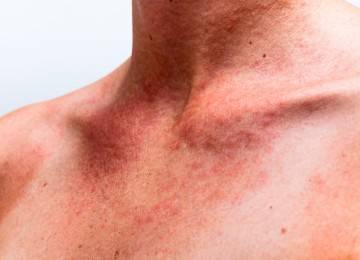
Клиническая картина синдрома
Обычно дети с синдромом Шерешевского-Тернера, рождаются раньше срока. Но даже в случае беременности, доношенной до 40 недель, вес новорожденного редко достигает 3 кг, а рост более 50 см.
Симптомы синдрома Шерешевского-Тернера
Сразу же после родов, можно заметить у ребенка признаки, которые характерны для заболевания:
- Укороченная шея;
- Птеригиум-синдром – кожные складки в виде крыльев на боках шеи;
- Нарушение оттока лимфы;
- Отеки на стопах и кистях;
- Наличие врожденных пороков сердечно-сосудистой системы.
Далее на первый план выступают проблемы с кормлением – у детей нарушено сосание, они часто срыгивают «фонтаном», находится в моторном возбуждении. На первых годах жизни можно заметить отставание в развитие – ребенок поздно начинает сидеть, ходить, говорить. Также для синдрома Шерешевского-Тернера характерно частое, повторное возникновение среднего отита, которое впоследствии приводит к кондуктивной тугоухости.
На момент полового созревания люди с синдромом Шерешевского-Тернера выглядят следующим образом: рост людей редко когда превышает 150 см. Месячные при синдроме Шерешевского-Тернера отсутствуют.
Такая болезнь, синдром Шерешевского-Тернера, имеет характерные фенотипические проявления.
Кроме этого, они имеют характерный внешний вид:
- Лицо обретает определенное выражение – «лицо Сфинкса»;
- Шея укорочена, присутствует птеригиум-синдром;
- Граница роста волос занижена;
- Челюсть недоразвита – микрогнатия;
- Уши увеличены, часто бывает лопоухость;
- Грудная клетка слишком широкая.
Жизнь с синдромом Шерешевского-Тернера непроста, при данной патологии значительно поражается костная система организма. Часто возникает сколиоз, дисплазия суставов, особенно тазобедренных, девиация локтей. На черепе наблюдается микрогнатия, неправильный прикус и готическое небо. В связи с недостаточным количеством эстрогена, люди с таким синдромом подвержены раннему возникновению остеопороза. У них часто случаются переломы позвоночника, костей кисти и шейки бедра.
Продолжительность жизни у людей с синдромом Шерешевского-Тернера
Сколько живут люди с синдромом Шерешевского-Тернера зависит от степени тяжести заболевания и от осложнений систем органов, которые возникают вследствие данной патологии. Со стороны сердечно-сосудистой системы встречаются такие пороки, как коарктация и аневризма аорты, дефекты межжелудочковой и межпредсердной перегородки. С такими пороками прогноз для жизни неблагоприятный. В почках часто бывает раздвоение лоханок, стеноз артерий, подковообразная форма самой почки. Подобные патологии приводят к артериальной гипертензии. У больных синдромом Шерешевского-Тернера нередко развивается косоглазие, близорукость и опущение века. К тому же они могут также страдать и дальтонизмом.
Умственные способности, как правило, сохраняются на должном уровне, но в некоторых случаях может наблюдаться олигофрения. Людей, страдающих синдромом Шерешевского-Тернера, часто сопровождают соматические заболевания – алопеция, микседема, витилиго, дефицит ферментов в тонком кишечнике, ожирение. Практически во всех случаях диагностируется диабет первого или второго типа и ишемическое поражение сердца. Доказано, что у таких пациентов рак толстого кишечника возникает в несколько раз чаще, чем у здоровых людей.
Характеристика женщин, страдающих данным заболеванием
Практически у 100% больных девушек диагностируется первичный гипогонадизм. Внутренние половые органы недоразвиты – матка гипоплазирована, а вместо яичников с двух сторон находятся фиброзные тяжи. Женщины с синдромом Шерешевского-Тернера обычно полностью стерильны – в яичниках отсутствуют фолликулы с яйцеклетками. Вульва видоизменена: большие губы похожи на мошонку, а малые и клитор недоразвиты. Девственная плева может отсутствовать. Некоторые задаются вопросом как приходят месячные с синдромом Шерешевского-Тернера — в период полового созревания обнаруживается первичная аменорея. Грудные железы развиты недостаточно, наблюдается незначительное оволосение в участке лобка и подмышек. Беременность, которая возникла природным путем, возможно только в случае мозаичной формы поражения X-хромосомы.
Мальчики при синдроме Шерешевского Тернера
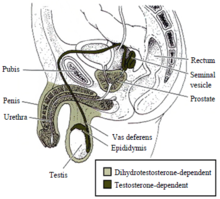
Половые органы мужчины также недоразвиты. Яички гипоплазированы, часто не опускаются в мошонку, иногда диагностируется анорхия – полное отсутствие ткани яичек в организме. Наблюдается сильно заниженный уровень мужского гормона – тестостерона.






![Синдром тёрнера (синдром шерешевского — тёрнера) [lifebio.wiki]](https://blotos.ru/wp-content/uploads/b/d/9/bd91127c6596bf6626e08f5aa44d8d62.jpg)
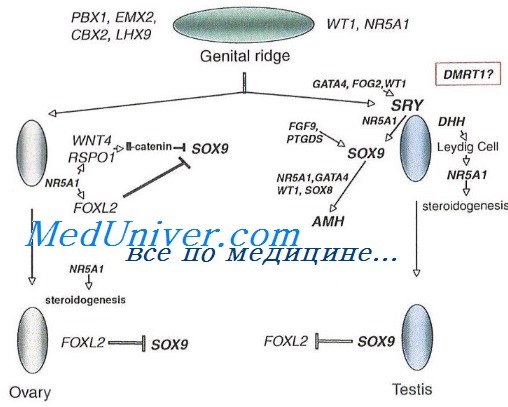


Патологическая анатомия
При Тернера синдроме гонады обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонады. Реже встречаются рудименты яичников (см.), включающие овариальную строму, а иногда и отдельные примордиальные фолликулы. Последние могут наблюдаться у больных с кариотипом 45,X/46,XX; 45,X/47,XXX. При кариотипе 45,X/46,XY в гонадных тяжах могут быть обнаружены элементы яичек (см. Яичко) — канальцы, клетки Лейдига (интерстициальные эндокриноциты) и Сертоли (поддерживающие клетки) и рудименты семявыносящего протока (см.).
Прочие патологоанатомические данные соответствуют особенностям клин, проявлений. Наиболее важны изменения костио-суставной системы — конкресценция позвонков (см. Позвоночник), spina bifida (см.), деформация суставов (см.), укорочение метакарпальных и метатарзальных костей, фаланг (см. Кисть, Стопа), остеопороз (см.) и др., изменения сердца (см.) и крупных сосудов — коарктация аорты (см.), незаращение боталлова протока (см. Артериальный проток), межжелудочковой перегородки, стеноз устья аорты, почечных артерий и др., пороки развития почек (см.) — подковообразная почка, удвоение лоханок и мочеточников и др.
лечение
В качестве условия хромосомного, нет никакого лечения синдрома Тернера. Тем не менее, многое можно сделать, чтобы свести к минимуму симптомы. Например:
Гормон роста , либо самостоятельно , либо с низкой дозой андрогена , увеличит рост и вероятно окончательного рост взрослого человека . Гормон роста одобрен США продовольствия и медикаментов для лечения синдрома Тернера и покрыт многими страховыми планами. Существует доказательство того, что это эффективно, даже у детей раннего возраста.
Эстроген заместительной терапии , такие как таблетки контроля рождаемости , была использована , так как условие было описано в 1938 году , чтобы способствовать развитию вторичных половых признаков. Эстрогены имеют решающее значение для поддержания хорошей целостности костей, сердечно — сосудистых заболеваний и здоровья тканей. Женщины с синдромом Тернера , которые не имеют спонтанной половой зрелости и, не получавших эстроген с высоким риском развития остеопороза и сердечных условиях.
- Современные репродуктивные технологии также были использованы , чтобы помочь женщинам с синдромом Тернера забеременеть , если они желают. Так , например, донор яйцеклетки могут быть использованы для создания эмбриона, который переносится на синдром Тернер женщиной.
- Маточная зрелость положительно связана с годами использование эстрогена, истории спонтанного менархе и отрицательно связана с отсутствием текущей заместительной гормональной терапии.
причина
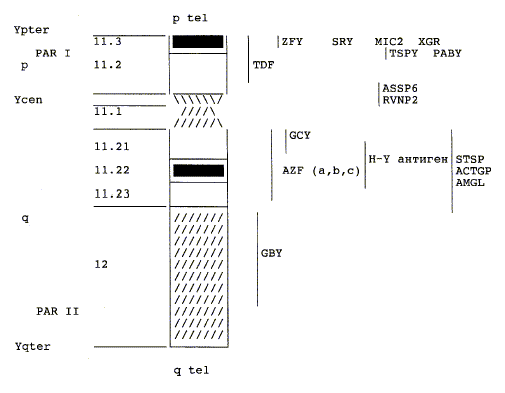
Синдром Тернера вызван отсутствием одного полной или частичной копии Х — хромосомы в некоторых или все ячейками. Эти аномальные клетки могут иметь только один X ( моносомии ) (45, X) , или они могут быть затронуты одним из нескольких типов как делеции короткого р плеча одного Х — хромосомы (46, X, дель (Хр) ) или наличие isochromosome с двумя руками (д 46, X, I (Xq)) синдром Тернера имеет определенные особенности в связи с отсутствием Псевдоаутосомной Области , которые , как правило , избавлены от X-инактивации. В мозаичных особей, клетки с X моносомии (45, X) может происходить наряду с клетками , которые являются нормальными (46, XX), клетки , которые имеют частичную monosomies, или клетки , которые имеют Y — хромосомы (46, XY). Наличие мозаицизма оценивается относительно распространенным в пораженных индивидуумов (67-90%).
наследование
В большинстве случаев , когда происходит моносомия, Х — хромосома происходит от матери. Это может быть связано с нерасхождением в отца. Мейотические ошибки , которые ведут к образованию X с р рычажных делеций или аномальных хромосом Y также встречается в основном у отца. Isochromosome Х или кольцевой хромосомой Х, с другой стороны образованы одинаково часто обоих родителей. В целом, функционал Х — хромосома обычно происходит от матери.
В большинстве случаев, синдром Тернера является спорадическим событием, и для родителей человека с синдромом Тернера риск рецидива не увеличивается при последующих беременностях. Редкие исключения могут включать в себя наличие сбалансированной транслокации Х — хромосомы в родительском, или там , где мать имеет 45, X мозаицизм ограничивается ее зародышевых клеток.
Яичниковая аменорея
Эта форма патологии наблюдается при преимущественном поражении яичников и относительно сохраненной функции гипофиза. Встречается она в 40-50% всех случаев аменорей (К. Н. Жмакин, 1966). В основе лежит полное или частичное отсутствие гормональной активности яичников. Клиническая картина заболевания зависит от времени появления и степени выраженности яичниковой недостаточности. В случаях врожденных дефектов или повреждения яичников в детском или пубертатном возрасте возникает первичная аменорея, которая сопровождается различными нарушениями сомато-по-лового развития и половой дифференцировки. Повреждения, наступившие после полового созревания, вызывают вторичную аменорею и не приводят к изменениям фенотипа. Если функция яичников частично сохраняется, периоды аменореи могут сменяться гипоменструальным синдромом или дисфункциональными кровотечениями.
Вторичная яичниковая аменорея, как правило, имеет гипо-гормональный характер и сопровождается резким снижением уровня обоих половых гормонов.
Врожденные формы первичной аменореи чаще всего являются результатом хромосомных аномалий.
Беременность при синдроме Шерешевского-Тернера
Когда на свет появляются детки с таким заболеванием, родители должны знать, что в этом никто из них не виноват, так как это нарушение возникает на уровне хромосом.
Обычно дети с данным синдромом рождаются у женщин, беременность которых сопровождалась тяжелым токсикозом, присутствовала угроза выкидыша или проходили преждевременные роды с отклонениями. Другой особенностью является хромосомная патология. Как правило, у плода первичные половые клетки закладываются с уже сформированным набором хромосом. Когда после 4-х месяцев начинается активное развитие плода, то оно происходит в обратном направлении, поэтому к рождению малыша количество стандартных половых гормонов сокращается. Такое явление может по-разному отражаться на ребенке, например, в виде половинного полового недоразвития или бесплодия.
Однако нередко женщины имеющие такое заболевание ведут полноценную супружескую жизнь. Кроме этого, возможно благоприятное течение заболевания, когда синдром обладает мозаичным характером, при котором у женщин присутствуют менструации, наступает беременность и возможен полноценный родовой процесс.
Диагностика синдрома Шерешевского-Тёрнера
У новорожденных диагноз можно заподозрить при наличии лимфедемы или крыловидной складки шеи. В отсутствие этих изменений диагноз у некоторых детей выявляют позже на основании низкого роста, аменореи и отсутствия пубертата. Диагноз подтверждается исследованием кариотипа. Для обнаружения врожденных пороков сердца показано проведение эхокардиографии или МРТ.










Цитогенетический анализ и исследования с Y-специфичным зондом проводят всем лицам с дисгенезией гонад для исключения мозаицизма с наличием клеточной линии с кариотипом 46, XY (45, X/46XY). У таких пациентов обычно женский фенотип с различными чертами синдрома Тернера. Они находятся в группе повышенного риска по развитию злокачественных новообразований гонад, особенно гонадобластомы, поэтому для профилактики сразу после определения диагноза следует удалить гонады.
[], [], [], [], [], []Физическое обследование
Диагноз ставят на основании характерной клинической картины: короткая шея с избытком кожи и крыловидными складками у новорождённых девочек, лимфатический отёк кистей и/или стоп, врождённые пороки левого сердца или аорты (особенно коарктация аорты), задержка роста и полового развития в пубертатный период у девочек.
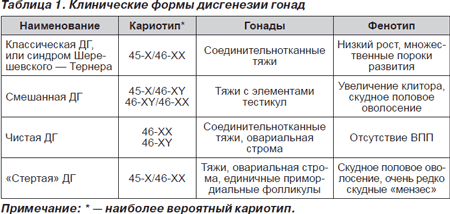
Что представляет собой дисгенезия гонад?
Эта патология является врожденной. Характерные признаки заключаются в недоразвитии или в отсутствии половых желез. Основная причина заболевания – хромосомные аномалии.
У больных людей встречается недобор хромосом, мозаицизм, дефект короткого плеча и другие явления. Из-за подавления мейоза женские яичники не могу полноценно развиваться, при этом яйцеклетки вообще исчезают ещё до момента рождения девочки.
Специалисты утверждают, что при дисгенезии гонад нарушаются гены, отвечающие за половое развитие, рост костей и мышц, сердечно-сосудистой и мочевой системы. Исходя из комбинации этих нарушений, различают несколько форм патологии, которые проявляются:
Х-хроматин-положительные варианты синдрома дисгенезии гонад
Характерные соматические клинические проявления, равно как и особенности состояния гонад, могут отмечаться у пациентов с аномалиями структуры Х-хромосомы (делеции или добавления) и мозаицизмом половой хромосомы на фоне набора 45, X. Факты свидетельствуют, что гены как длинного, так и короткого плеч Х-хромосомы определяют половую дифференцировку, тогда как гены короткого плеча Х-хромосомы преимущественно предупреждают развитие низкорослости и соматических нарушений, наблюдаемых у пациентов 45, X. Обычно у пациентов с 45, Х/46, XX мозаицизмом имеется нормальный (а не характерный для 45, X) фенотип и даже может отмечаться нормальное половое развитие и фертильность. У пациентов с дупликацией длинного плеча Х-хромосомы и делецией короткого плеча — с так называемой Xq-изохромосомой — отмечается увеличение частоты развития аутоиммунного тиреодита, сахарного диабета 2-го типа и воспалительных заболеваний кишечника по сравнению с пациентами 45, X. У пациентов с кариотипом 46, Хг (X) может быть отставание в умственном развитии и пороки развития, не всегда связанные с синдромом Тернера. Эти нарушения связаны с недостатком инактивации малого кольца Х-хромосомы и, следовательно, функциональной дисомией генов кольцевой и нормальной Х-хромосом.
Симптомы синдрома Шерешевского-Тёрнера
У многих новорожденных отмечаются только очень легкие проявления; однако у некоторых наблюдаются выраженная дорсальная лимфедема кистей рук и ступней, а также лимфедема или кожные складки на задней поверхности шеи. Другие распространенные аномалии включают крыловидные складки шеи, широкую грудную клетку и втянутые соски. У пораженных девочек отмечается низкий рост по сравнению с членами семьи. Менее частыми признаками являются низкая линия роста волос на задней поверхности шеи, птоз, множественные пигментные невусы, короткие четвертые пястная и плюсневая кости, выступающие подушечки пальцев с завитками на концах пальцев, а также гипоплазия ногтей. Также отмечаются cubitus valgus (вальгусное отклонение в локтевом суставе).
Распространенные аномалии сердца включают коарктацию аорты и двухстворчатый клапан аорты. Часто с возрастом развивается гипертензия, даже в отсутствие коарктации. Нередко встречаются аномалии почек и гемангиомы. Иногда в ЖКТ обнаруживают телеангиэктазию, с развивающимися желудочнокишечными кровотечениями или потерей белка.
Дисгенезия гонад (яичники замещаются двухсторонними тяжами фиброзной стромы с отсутствием развивающихся половых клеток) отмечается у 90 % больных, приводя к отсутствию пубертата, отсутствию увеличения грудных желез, аменорее. В то же время у 5-10 % пораженных девочек спонтанно происходит менархе, и очень редко пораженные женщины фертильны и имеют детей.
Задержка умственного развития отмечается редко, но у многих пациентов наблюдается снижение некоторых перцептивных возможностей и вследствие этого низкие оценки в невербальных тестах и по математике, даже несмотря на то, что баллы, полученные за вербальный компонент тестов на интеллект, средние или даже высокие.
Диагностика
Пренатальная
Синдром Тернера может быть диагностирован во время беременности с помощью амниоцентеза или биопсии ворсин хориона.
Как правило, плод с синдромом Тернера может быть выявлен посредством отклоняющихся от нормы результатов ультразвукового исследования (например, порок сердца, аномалия почек, шейная гигрома, асцит). В исследовании 19 европейских журналов учета, 67,2% пренатально диагностированных случаев синдрома Тернера были обнаружены за счет аномалий при ультразвуковом исследовании. В 69,1% случаев была представлена одна аномалия, а в 30,9% случаев – две и более аномалии.
Повышенный риск развития синдрома Тернера также может быть выявлен с помощью отклоняющихся от нормы триады или квадруплета в обследовании материнской крови. Плод, диагностированный посредством положительного анализа материнской сыворотки, чаще имеет мозаичный кариотип, чем тот, диагноз которого основан на аномалиях ультразвукового исследования, и, наоборот, субъекты с мозаичными кариотипами менее вероятно имеют связанные ультразвуковые аномалии.
Хотя риск рецидива не повышается, семьям, в которых имеется случай беременности или ребенка с синдромом Тернера, часто рекомендуется генетическое консультирование.
Постнатальная
Синдром Тернера может быть диагностирован постнатально в любом возрасте. Часто он диагностируется при рождении в связи с проблемами с сердцем, необычно широкой шеей и отеком рук и стоп. Тем не менее, часто встречается, что он остается недиагностированным в течение нескольких лет, обычно пока девочка не достигнет возраста полового созревания/пубертатного возраста и не сможет начать развиваться надлежащим образом (изменения, связанные с половым созреванием, не произойдут). В детстве о синдроме Тернера может свидетельствовать низкий рост.
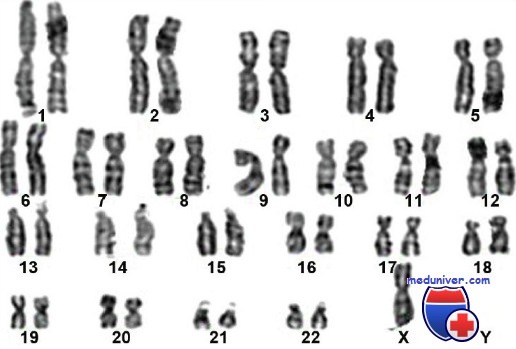
Анализ, носящий название кариотип или анализ хромосом, анализирует хромосомный состав субъекта. Это предпочтительный анализ с целью диагностики синдрома Тернера.
Механизм развития

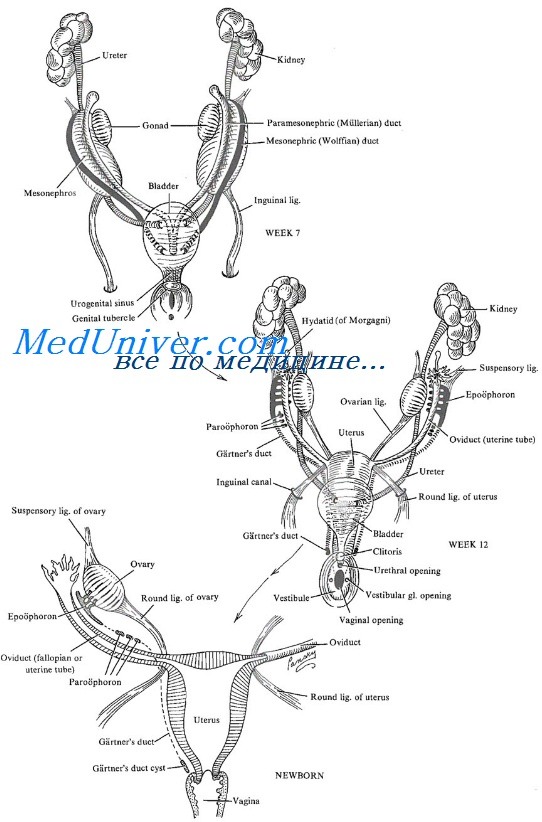
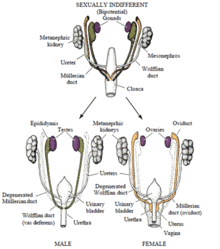
Развитие дисгенезии происходит на стадии эмбриогенеза. Формирование основных половых признаков заканчивается на 5-6 неделе беременности. В течение всего первого триместра возможны отклонения в фенотипе будущего ребенка.






![Синдром тёрнера (синдром шерешевского — тёрнера) [lifebio.wiki]](https://blotos.ru/wp-content/uploads/d/b/6/db67d9953edeb50e46f110ea56280633.jpg)

Для адекватного формирования желез требуется две хромосомы (XX или XY, яичник или тестикула соответственно). Развитие дисгенезии сопряжено с дефектом X-хромосомы (зачастую). Дифференциация желез не завершается. Возможно формирование нескольких форм патологического отклонения. В отличие от истинного гермафродитизма, полностью развиваются половые железы, присущие одному полу. Чаще женскому. Тестикулярный компонент присутствует в форме недифференцированного гонадального тяжа. Примерно в 20% случаев наблюдается полное отсутствие полноценных гонад, что ассоциировано с бесплодием.
Клинические признаки
Чаще всего у больных наблюдается мозаичная форма X/46, XY.
Насколько широко распространена данная патология неизвестно. Но согласно словам специалистов, она является одной из главных причин амбисексуальности половых органов у новорожденных, наряду с болезнью, которая связана с недостаточной выработкой кортизола надпочечниками.
В подавляющем большинстве случаев специалисты причисляют новорожденных с наличием смешанной дисгенезии к девочкам.
Развитие молочных желез происходит довольно редко. Основное число больных воспитываются как девочки.
Большинство фенотипических мальчиков остаются не полностью вирилизированными. В основном детородные органы у них амбисексуальны, в том числе немного увеличенный пенис, и половые губы, которые сращены в мошонку.
Практически всегда имеется вагина, матка и маточные трубы. Яички чаще находятся в брюшной полости, но иногда в мошонке или в каналах паха (в этом случае больного считают мальчиком).
До начала полового созревания, яичко выглядит вполне естественно. В постпубертатном периоде оно имеет большое количество созревших клеток Лейдига. Но в этом случае специалисты указывают на то, что зародышевых элементов в семенных канальцах нет и там можно обнаружить исключительно клетки Сертоли.
Период полового созревания отличается выработкой яичком необходимых андрогенов. В итоге пенис становится больше и проявляется типичная вирилизация. Феминизация при этом очень низкая, она преимущественно обусловлена выработкой эстрогенов. Данный процесс возникает, как правило, вследствие опухоли гонад.
Почти у тридцати процентов заболевших наблюдаются следующие признаки:
- Пониженная задняя линия оволосения;
- Грудная клетка, имеющая форму щита;
- Большое число окрашенных родимых пятен;
- Низкий рост (около ста пятидесяти сантиметров);
- Складчатость шеи;
- Неправильно развитые предплечья.
В основном половой хроматин у пациентов отрицательный. При обследовании было выявлено, что шестьдесят процентов из них обладают кариотипом 45, X/46, XY.
Остальные больные принадлежат к кариотипу 46, XY, частота мозаицизма бывает пониженной либо ограниченной некоторыми клеточными линиями. Мозаичную форму можно объяснить потерей Y-хромосомы на начальном периоде деления xy-зиготы. При этом осуществляется разделение тестикул, а потеря y-хромосомы оказывает воздействие на созревание строений из генитальных гребней. Но этот процесс до конца не завершается.
Необходимо отметить, что в некоторых ситуациях у больных разного возраста начинается развитие опухоли гонад. Появление опухоли наблюдается примерно у четверти пациентов. При этом большая часть состоит из семином а гонадобластомы встречаются не часто. Эти образования, по словам врачей, могут быть найдены еще до начала полового созревания.
Чаще всего с таким явлением сталкиваются больные с женским фенотипом. У них не обнаруживают каких-либо соматических симптомов, которые характерны для 45, X-патологии созревания яичников. Специалисты отмечают поражение внутрибрюшных яичек, гонадальные тяжи подвергаются поражению значительно меньше.
Лечение
Проводится под руководством целого «консилиума» специалистов: эндокринолога, гинеколога, генетика, хирурга и прочих.
Терапия начинается после диагностики состояния.
Основу этиотропного лечения составляет применение гормональных препаратов: эстрогенов для повышения выраженности половой принадлежности и дальнейшего развития желез, тиреоидных гормонов (стимуляция щитовидной железы), приостановка избыточного роста костей и структур опорно-двигательного аппарата или восстановление нормального развития (стероидные гормональные препараты).
Дополнительно показана длительная работа сначала с психотерапевтом, затем с психологом для нормализации эмоционально-волевого, психиатрического компонента и адаптации в социуме.
Коррекция пороков сердца, выделительной системы, церебральных структур проводится под контролем кардиологов и профильных хирургов, нефрологов, урологов, неврологов и нейрохирургов. Возможно назначение операции для устранения дефектов.





В течение всей жизни два раза в год рекомендуется проходить УЗИ органов малого таза (о данном методе диагностики мы уже рассказывали) и консультироваться с гинекологом для ранней диагностики онкологических патологий, к которым пациенты с дисгенезией гонад более склонны.
Лечение дисгенезии гонад проводится на протяжении всей жизни пациентки. Поддерживающая терапия предполагает применение гормональных средств.
Синдром Шерешевского-Тёрнера: лечение и диагностика
Главные задачи лечения людей, страдающих от заболевания — синдром Шерешевского-Тёрнера –профилактика осложнений, активная стимуляция роста, достижение нормальной регулярной менструации, половых вторичных половых признаков. Лечение может проводиться с ранних лет пациентки, но оно является достаточно неспецифичным. Так, для улучшения состояния пациенток используются:
- ЛФК;
- целебный массаж;
- полноценное питание;
- витаминные комплексы;
- охранительный режим.
Чтобы увеличить конечный рост девочки, специалист прописывает соматотропин – этот медпрепарат является рекомбинантным гормоном роста. Выполняются каждый день подкожные уколы, пока костный возраст пациентки не достигает пятнадцати лет, а скорость роста не уменьшится до двух сантиметров в год. Чаще всего ростостимулирующая терапия дает возможность женщинам вырасти до 155см, не больше. При этом лечиться гормоном роста они могут вместе с терапией различными анаболическими стероидами.
Чтобы имитировать стандартное половое созревание, заместительная терапия при помощи эстрогенов начинается с 14 лет пациентки. Приблизительно через 15-17 мес. вводится циклическая терапия, выполняется с использованием оральных прогестагенных контрацептивов. Необходимо отметить, что заместительная терапия гормонами должна выполняться до возраста естественной менопаузы, которая возникает где-то в пятьдесят лет у здоровых женщин. Если мужчины болеют синдромом, им рекомендуется ЗГТ половыми гормонами (мужскими).
Когда имеются гемодинамически значимые ВПС, производится их коррекция при помощи хирургических методов. Она позволяет устранить крыловидные складки шейки при помощи современных методик пластической хирургии.
Когда женщина достигла оптимального уровня собственного полового развития, она даже при наличии синдрома сможет иметь детей. Для этого будет задействоваться процедура ЭКО с применением донорской яйцеклетки. В случае наличия небольшой активности яичников могут для оплодотворения применяться собственные ооциты. Если наблюдается проблема сильного роста волос, решить ее можно при помощи эпиляции.

Профилактические мероприятия, прогнозы
В общем, синдром не оказывает заметного воздействия на продолжительность жизни пациента. Правда, имеются исключения, к которым относятся ситуации, связанные с тяжелыми ВПС, декомпенсацией сопутствующих болезней, ранним развитием и прочее. В случае наличия адекватной терапии люди, страдающие от синдрома, могут свободно создавать семьи, вступать с партнерами в интимную близость, но большинство из них все-таки являются бесплодными.
Если брать во внимание разнообразие проявлений данного синдрома, тогда введение/наблюдение пациентов должно выполняться медицинскими работниками сферы эндокринологии, генетики, педиатрии, андрологии, гинекологии, офтальмологии, кардиологии и прочее. Методики профилактики для рождения здоровых детей достаточно ограничены – это может быть медико-генетическая консультация, а также пренатальная диагностика
Особенности диагностирования
Синдром у новорожденных деток иногда диагностируется педиатром, неонатологом в том случае, когда имеется крыловидная складка шеи и, естественно, лимфедемы. В случае отсутствия подобных признаков достаточно часто диагноз можно поставить исключительно в пубертатном периоде. Тогда среди основных симптомов болезни:
- отсутствия менархе;
- плохой рост;
- вторичные половые признаки не выражены.
Количество гормонов определяется повышением гонадотропинов, а также значительным сокращением в крови количества эстрогенов. Как правило, значение при обследовании на наличие синдрома имеет выявление полого хроматина, эффективного исследования кариотипа. Если обнаружены специфические признаки заболевания у плода после проведенного акушерского ультразвукового исследования, тогда должен быть решен вопрос относительно проведения специального инвазивного диагностирования – пренатального.
Проводится дифференциальная диагностика и с гипофизарным нанизмом. Для этого следует провести исследование количества гормонов гипофиза, имеющихся в крови, а также электроэнцефалография. После осмотра и опроса врач назначает пациентке различные диагностические процедуры и анализы, которые помогут раскрыть общую картину болезни и назначить оптимальное лечение.








Перспективы беременности

Возможно ли наступление беременности при дисгенезии гонад? Сохранение фертильности и репродуктивной функции зависит от формы.
Типичная характеризуется возможностью случайной беременности. Количество яйцеклеток минимально, что обуславливает малые шансы на подобный исход. Чистая определяется теми же чертами.
При смешанной, вероятность наступления беременности максимальна. Овариальный резерв определяет короткий репродуктивный период: 3-6 лет. Спустя указанный срок наступает затухание фертильности — менопауза.
Репродуктивная функция при дисгенезии гонад существенно снижается, но наступление беременности все еще возможно. Усилия для реализации фертильности предполагают применение вспомогательных репродуктивных технологий.
Течение беременности почти во всех случаях сложное, требует постоянного контроля со стороны акушера-гинеколога.