Первичные дефициты клеточного иммунитета
К первичным дефицитам клеточного иммунитета относятся следующие заболевания:
- Синдром Ди Джорджи
- Синдром Дункана
- Недостаточность пуриннуклеозидфосфорилазы
- Оротацидурия
- Биотин-зависимые ферментопатии.
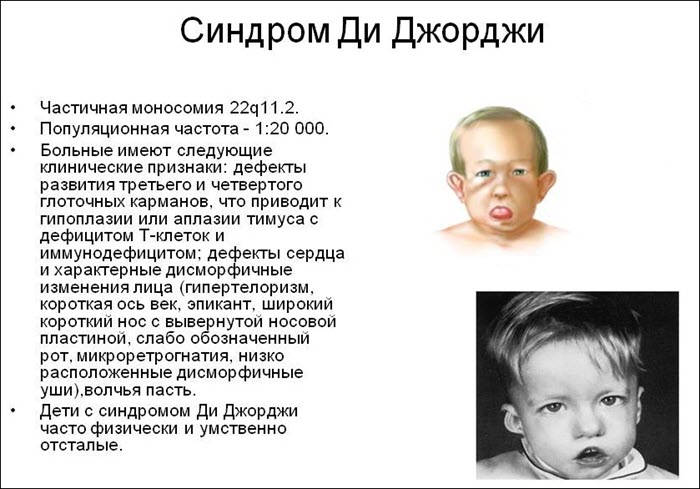
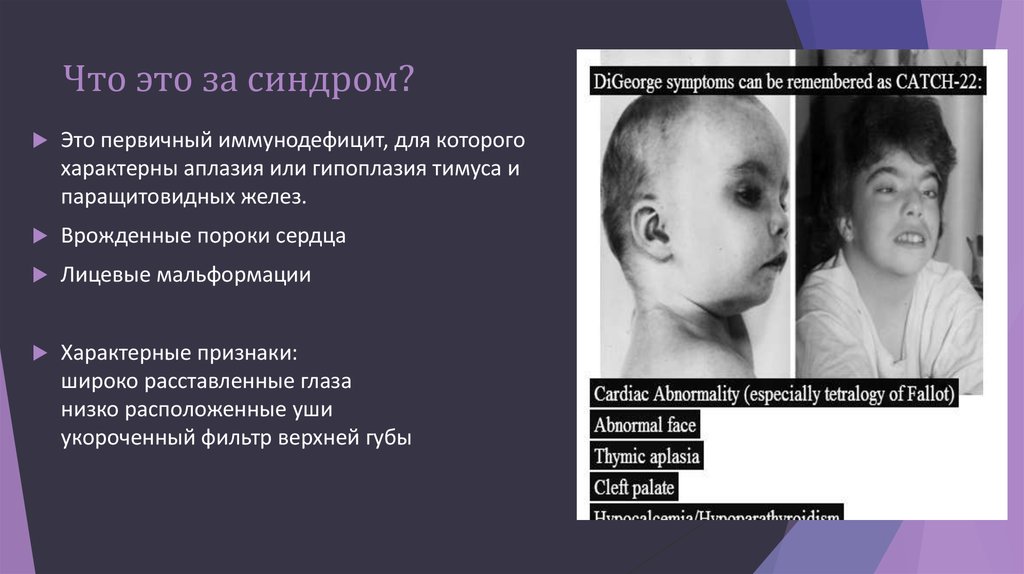
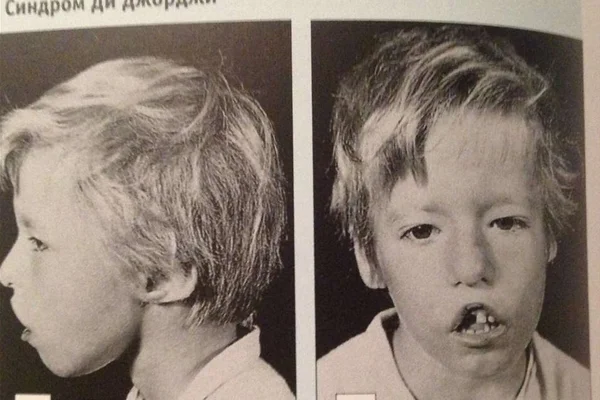
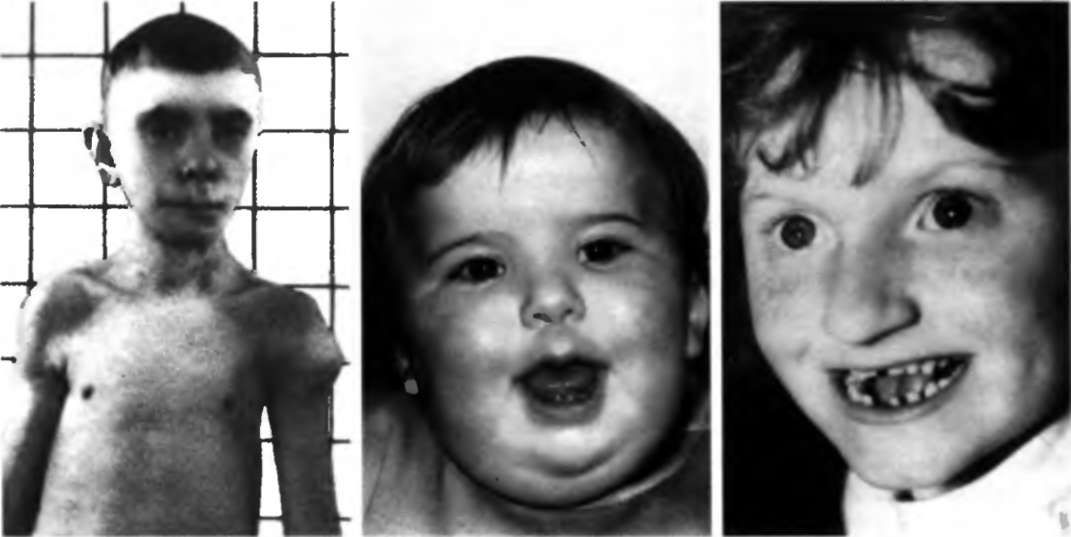
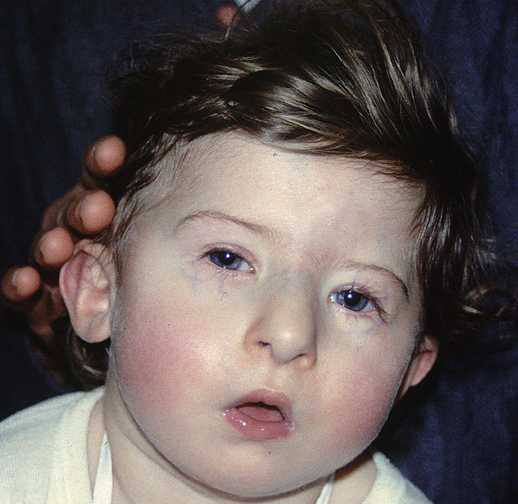
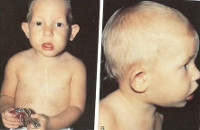
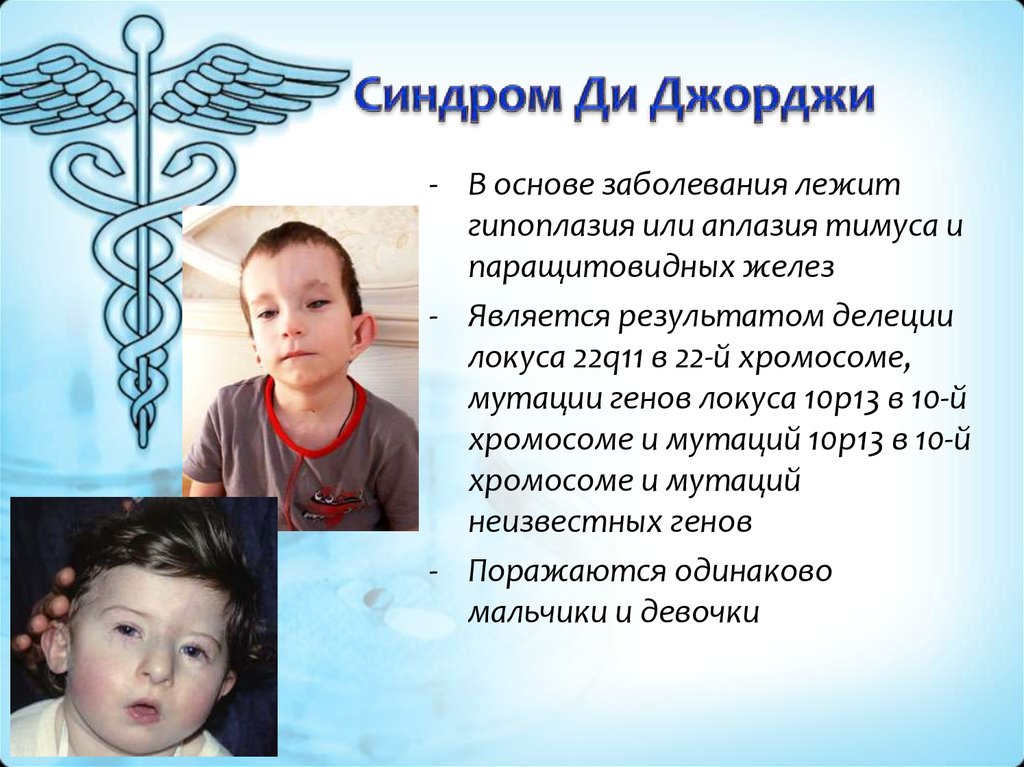
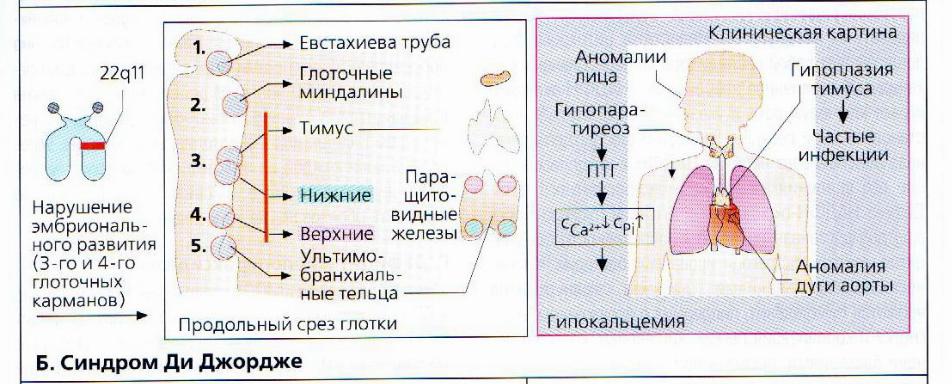
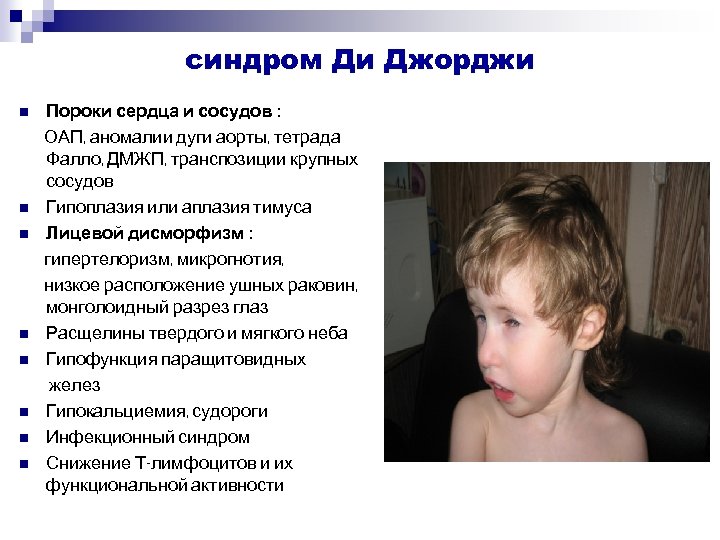
Синдром Ди Джорджи
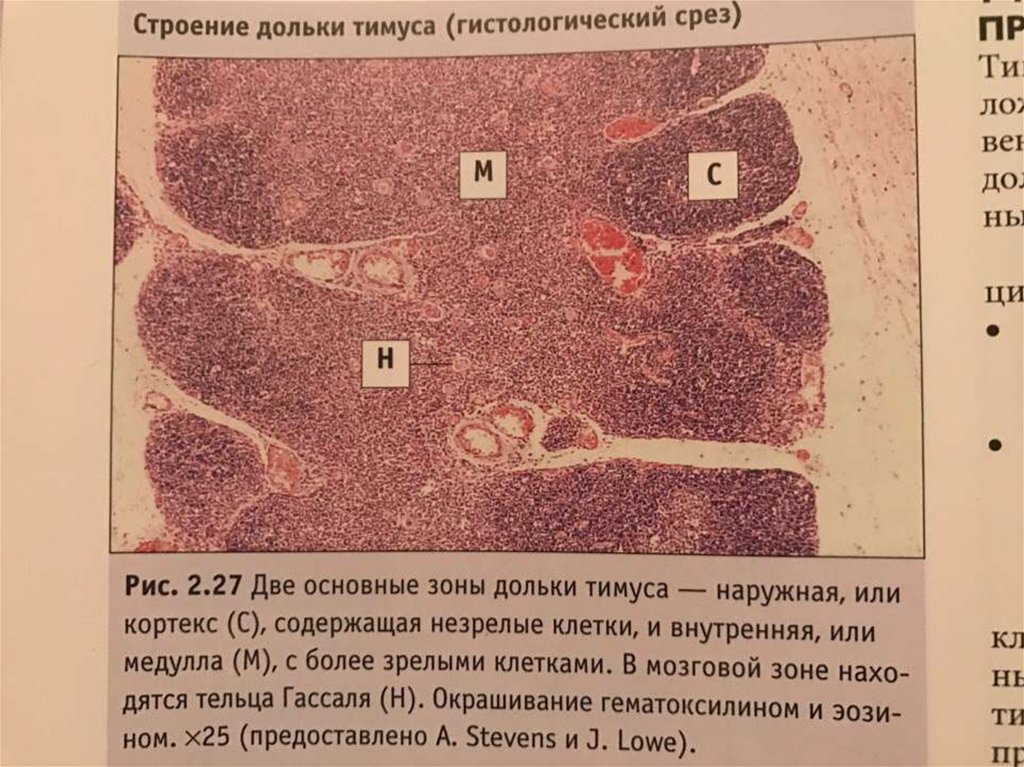
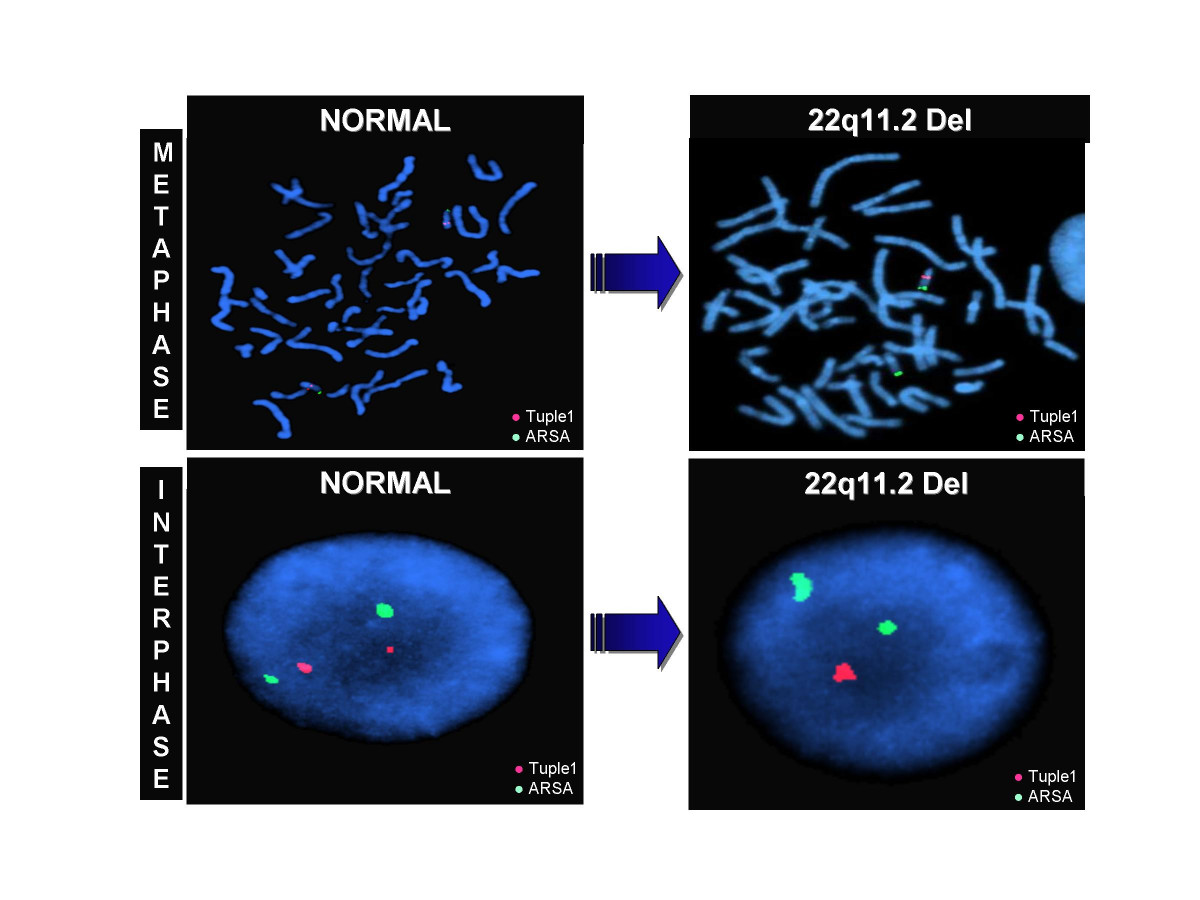
В основе синдрома Ди Джо́рджи (Di George) лежит гипоплазия тимуса. Синдром описан в г. Считается, что это заболевание не является наследственным, оно возникает в результате приобретённого нарушения органогенеза в области III—V жаберных дуг (глоточных карманов) на 6—8 неделе беременности. Поэтому, кроме порока тимуса, отмечаются дефекты околощитовидных желёз, сердца и крупных сосудов, а также орофациальные пороки (микростомия, микрогнатия, гипертелоризм, низкое расположение ушных раковин).
Результатом гипоплазии паращитовидных желёз является дефицит парат-гормона и персистирующая гипокальциемия, вследствие чего развивается судорожный синдром, который может проявиться уже в первые часы жизни (неонатальная тетания). Причиной смерти детей в более старшем возрасте служат осложнения, связанные с пороками развития сердца.
Нарушения, затрагивающие Т-лимфоциты, могут быть как очень глубокими, так и едва заметными. В любом случае функция Т-клеток с возрастом восстанавливается и к 5 годам, если ребёнок остаётся жив, не удаётся обнаружить их недостаточности. Антиген-независимый этап созревания Т-клеток при этом происходит вне тимуса — в многослойных плоских эпителиях, прежде всего в эпидермисе. Одним из эффективных способов лечения синдрома Ди Джорджи является трансплантация эмбриональной ткани тимуса.
Синдром Дункана
Синдром Ду́нкана (Х-сцепленный лимфопролиферативный синдром) — иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна—Барр. Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз, развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна—Барр.
Недостаточность пурин-нуклеозид-фосфорилазы
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
Оротацидурия
Оротацидури́я — наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты (оротата) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы, которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат, необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы, участвующих в метаболизме аминокислот с разветвлённой цепью — валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.
Классификация
Количество видов первичных иммунодефицитов достаточно велико. Это объясняется сложностью иммунной системы и тесной интеграцией ее отдельных звеньев, в результате чего нарушение работы или «выключение» одной части способствует ослаблению всей защиты организма в целом. На сегодняшний день разработана сложная разветвленная классификация подобных состояний. Она состоит из пяти главных групп иммунодефицитов, каждая из которых включает несколько наиболее распространенных типов патологии. В упрощенном варианте данную классификацию можно представить следующим образом:
- Первичные дефициты клеточного иммунитета. Группа объединяет состояния, обусловленные недостаточной активностью или низким уровнем Т-лимфоцитов. Причиной может выступать недостаточность тимуса, ферментопатии и иные (преимущественно генетические) нарушения. Наиболее распространенными формами иммунодефицитов такого типа являются синдромы Ди Джорджи и Дункана, оротацидурия, недостаточность ферментов лимфоцитов.
- Первичные дефициты гуморального иммунитета. Группа состояний, при которых понижена функция преимущественно В-лимфоцитов, нарушен синтез иммуноглобулинов. Большинство форм относится к категории дисгаммаглобулинемий. Наиболее известны синдромы Брутона, Веста, дефициты IgM или транскобаламина II.
- Комбинированные первичные иммунодефициты. Обширная группа заболеваний с пониженной активностью как клеточных, так и гуморальных звеньев иммунитета. По некоторым данным, этот тип включает более половины всех разновидностей иммунной недостаточности. Среди них выделяют тяжелые (синдром Гланцманна-Риникера), умеренные (болезнь Луи-Бар, аутоиммунный лимфопролиферативный синдром) и минорные иммунодефициты.
- Первичная недостаточность фагоцитов. Генетические патологии, вызывающие пониженную активность макро- и микрофагов – моноцитов и гранулоцитов. Все заболевания этого типа разделяются на две большие группы – нейтропении и дефекты активности и хемотаксиса лейкоцитов. Примерами являются нейтропения Костмана, синдром «ленивых лейкоцитов».
- Дефициты белков комплемента. Группа иммунодефицитных состояний, развитие которых обусловлено мутациями генов, кодирующих компоненты комплемента. В результате нарушается образование мембраноатакующего комплекса, страдают другие функции, в которых участвуют данные белки. Это вызывает комплемент-зависимые первичные иммунодефициты, аутоиммунные состояния или наследственный ангионевротический отек.
Профилактика
К сожалению, первичный иммунодефицит не может быть рассмотрен с точки зрения его профилактики. Это объясняется его врожденным характером. Здесь не требуются дальнейшие разъяснения. Судить о профилактике можно только в том случае, если рассматривают вторичный иммунодефицит. В этой ситуации нужно помнить о защищенном сексе и об использовании стерильных медицинских средств. Также правила личной гигиены должны стоять в приоритетах. Каждый может хотя бы защититься от вторичного иммунодефицита.
Первичный иммунодефицит может только поддерживаться специально подобранной в индивидуальном порядке терапией. В самых запущенных и тяжелых случаях применяют мощные иммуностимуляторы. Однако назначить подобного рода средства может только специалист высокой квалификации. Ни о каком самолечении не может идти речи!
Иногда используют цитокины, чтобы повысить функции лимфоцитов, а иногда и требуется пересадка костного мозга. Все сугубо индивидуально
Поэтому важно рассматривать ситуацию после взятых анализов. Обязательным является обращение к врачу иммунологу
Конечно, поддерживать организм и проводить своеобразное лечение лучше как можно раньше. Однако даже в запущенных ситуациях нужно ориентироваться на профессиональную помощь.


Иммунологические синдромы
Иммунологические синдромыИтак, выделяется 6 иммунопатологических синдромов:1. Инфекционный синдром.2. Аллергический синдром.3. Аутоиммунный синдром.4. Первичный иммунодефицит.5. Вторичный иммунодефицит.6.Имунопролиферативний синдром.
Инфекционный синдром
Инфекционный синдром – для него наиболее характерны:1. длительный субфебрилитет, лихорадка нечеткой этиологии;2. хронические инфекции ЛОР– органов (синуситы, отиты), лимфадениты;3. хронические заболевания дыхательных путей, которые часто повторяются;4. высокая частота острых респираторных вирусных заболеваний (у взрослых более чем 4 раза, у детей – более 6 раз в год);5. бактериальные поражения кожи и подкожной клетчатки (пиодермии, фурункулез, абсцессы, флегмоны, рецидивирующие парапроктиты у взрослых);6. грибковые поражения кожи, слизистых оболочек и ногтей;7. паразитарные инфекции;8. афтозные стоматиты, заболевания пародонта, кариес;9. рецидивирующие гнойные конъюнктивиты;10. рецидивирующий герпес различной локализации;11. повторные лимфадениты;12. хронические урогенитальные инфекции (хронический гнойный вульвит, уретрит, рецидивирующий цистит и пиелонефрит);13. дисбактериоз кишечника, хроническая гастроэнтеропатия с диареей неизвестного генеза;14. генерализованные инфекции.
Аллергический синдром
Наиболее характерными его проявлениями являются:1. аллергические заболевания кожи (атопiчний и контактный дерматит, крапивница, отек Квинке, феномен Артюса, экзема);2. аллергические заболевания ЛОР – органов (полiноз, хронический аллергический ринит и риносусит);3. бронхиальная астма;4. признаки пищевой аллергии (непереносимость пищевых продуктов);5. признаки лекарственной аллергии;6. признаки непереносимости химических соединений.
Аутоиммунный синдром
1. воспалительные заболевания опорно – двигательного аппарата (ревматоидный артрит);2. системная красная волчанка, дерматомиозит, склеродермия;3. системные васкулиты (гранулематоз Вегенера, узловатый периартериит и др.);4. гломерулонефриты;5. патология щитовидной железы, инсулинозависимый сахарный диабет, болезнь Аддисона, синдром Шегрена и другие гормональные нарушения);6. неврологические заболевания (рассеянный склероз, миастения Гравис и др..)7. неспецифический язвенный колит;8. аутоиммунные заболевания печени;9. аутоиммунные формы бесплодия, патология беременности, тяжелые формы течения климактерического синдрома;10. некоторые виды психопатологии (шизофрения).
Первичные иммунодефициты (преимущественно у детей)
1. Синдром Луи – Бар – атаксия в сочетании с телеангиэктазии, пятнами гипер – и депигментации;2. Синдром Вискота – Олдрича – геморрагический симптомокомплекс в сочетании с экземой и тромбоцитопенией у мальчиков;3. синдром Ди–Джорджи – судорожный синдром с гипокальциемией, пороками развития лицевого скелета и сердечно – сосудистой системы, гипоплазией тимуса;4. наследственные ангионевротический отек различной локализации (недостаточность ингибитора С1 компонента комплемента).
Вторичные иммунодефициты
1. все виды инфекционного синдрома в случае длительного торпидного к терапии лечения, тенденции к генерализации процесса;2. алопеция, де– и гиперпигментация кожи;3. СПИД;4.другие случаи приобретенной иммунной недостаточности.
Лимфопролиферативный синдром
1. опухоли в иммунной системе (лимфолейкозы, лимфосаркомы, болезнь Ходжкина, лимфомы, саркома Капоши);2. Х – сжатых рецессивный лимфопролиферативный синдром у детей;3. гиперплазия всех групп лимфатических узлов с воспалительными процессами в них в сочетании с частыми бактериальными инфекциями другой локализации;4. спленомегалия;5. мононуклеоз в анамнезе.
Таким образом, после опроса больного, мы можем заподозрить вероятный иммунологический синдром и направить больного для дальнейшего лабораторного исследования с определенным перечнем иммунологических тестов.
Симптоматика
Первые симптомы патологии появляются сразу после рождения ребенка. Восстановление Т-клеточного иммунитета наблюдается у детей, переживших 6-месячный возраст.

Клинические признаки синдрома:
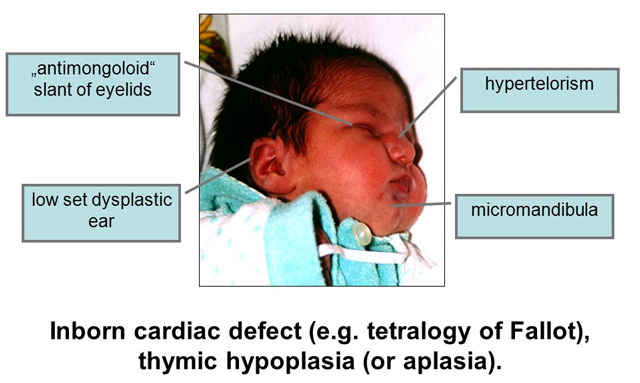
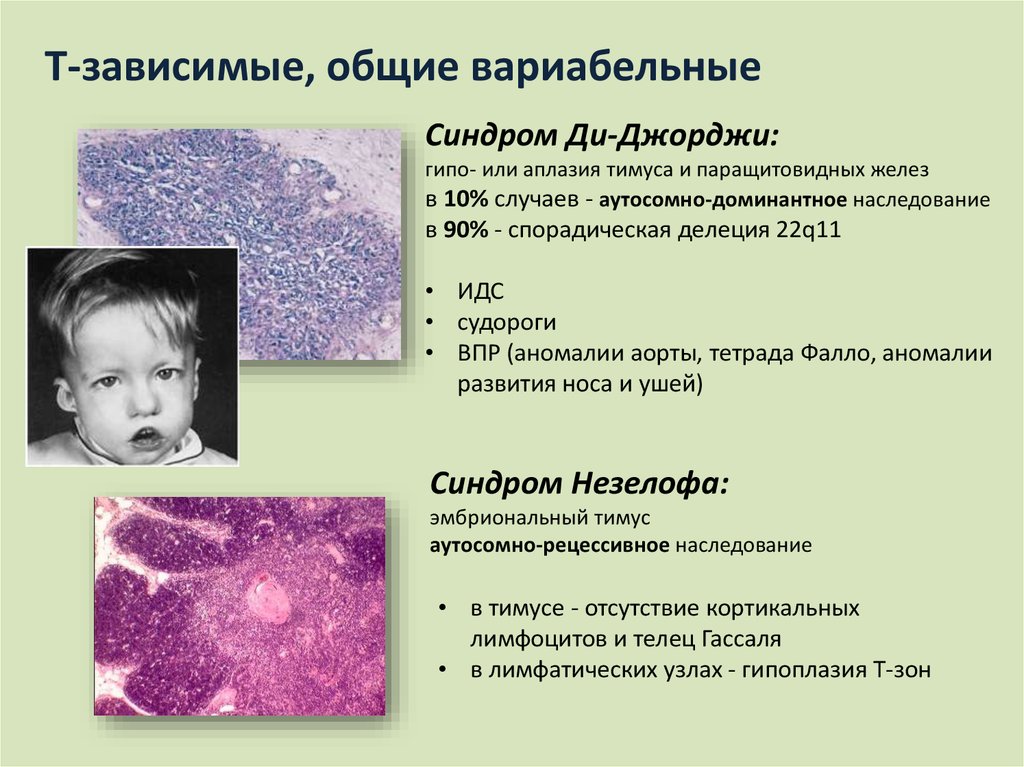
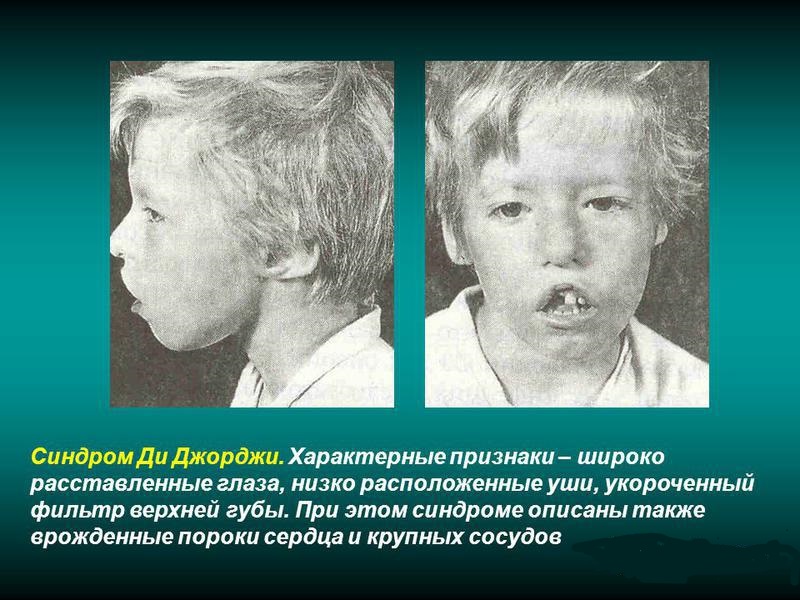
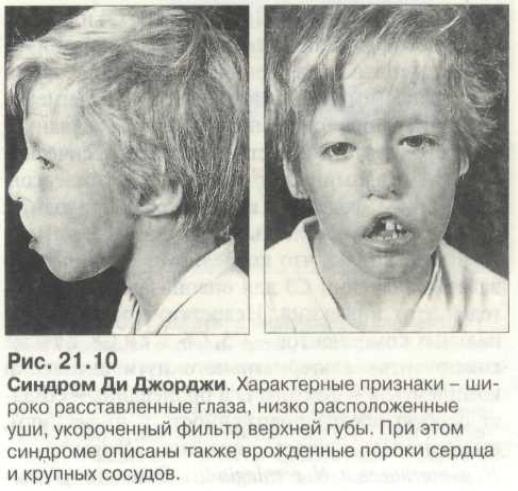
- Невооруженным глазом можно обнаружить аномалии лица, к которым относятся: расщепление неба, “готическое» небо, микрогнатия верхнечелюстных костей, «рыбий» рот, маленький нос с широкой переносицей, деформированные и низко расположенные ушные раковины, микроцефалия, широко расставленные глаза, косоглазие, наличие эпиканта, специфический разрез глаз с опущением наружных уголков.
- Проявления врожденных пороков сердца также выступают на первый план. У больных появляются признаки сердечной недостаточности: акроцианоз, тахикардия, одышка после физической нагрузки. Подобные процессы в организме больного ребенка требуют оказания квалифицированной медицинской помощи. В противном случае может развиться сердечная недостаточность или наступить ранняя смерть.
- Гипоплазия паращитовидных желез приводит к гипокальциемии и появлению у детей судорог и тетании, которые возникают при снижении концентрации в крови паратгормона. Судорожный синдром гипокальциемического типа возникает в первые дни после рождения ребенка, не купируется противосудорожными препаратами и нередко приводит к смерти младенца.
- Первичный иммунодефицит – следствие гипоплазии вилочковой железы. Ослабление естественной резистентности организма приводит к затяжным и тяжелым инфекционным заболеваниям, плохо поддающимся стандартной противомикробной терапии. У больных детей обнаруживается явная тенденция к инфекциям верхних дыхательных путей, протекающим в виде ринофарингита, отита, бронхопневмонии; дигестивным и кожным инфекциям, протекающим в виде диареи и пиодермии соответственно.
- При поражении ЦНС частично атрофируется кора головного мозга, возникает гипоплазия мозжечка, что проявляется нарушением походки, парезами и параличами, изменением чувствительности. Возможно у больных развитие умственной отсталости и возникновение неврологических расстройств, которые проявляются у детей с первых дней жизни. У ребят постарше отмечается беспокойство и психоэмоциональная лабильность. Психиатрическая патология у подростков проявляется синдромом гиперактивности, шизофренией, маниакально-депрессивным психозом.
- Аномалии пищеварительной системы – укороченный пищевод, отсутствие ануса; дыхательной системы – укорочение и сужение гортани, глотки, трахеи.
- Патология глаз проявляется изменением передней камеры глаза, колобомой, аномалией сосудистой оболочки и сетчатки.
- Со стороны мочевыделительной системы возникают следующие изменения: гидронефроз, атрофия почек, рефлюкс мочи в почечные чашечки и лоханки.
- Поражение костной системы включает аномалии скелета и зубов. Больные рождаются с полидактилией и отсутствием ногтей. У них поздно прорезываются зубы, возникают спонтанные переломы костей, нарушается правильное развитие зубной эмали, развивается кариес.
- Среди прочих проявлений синдрома выделяют: ларингомаляцию, трахеомаляцию, гастроэзофагальный рефлюкс, глухоту, нарушение глотания, паховые грыжи.
Оппортунистические инфекции редко угрожают жизни детей с синдромом Ди Джорджи. Обычно у них возникают рецидивирующие отиты и синуситы, обусловленные не только снижением иммунной защиты, но и аномальным строением лицевого скелета.




У пациентов с синдромом Ди Джорджи в крови появляются Т-клетки, обладающие аутоагрессией, что проявляется развитием аутоиммунных заболеваний – цитопении, аутоиммунного тиреоидита, ювенильного ревматоидного артрита, аутоиммунной гемолитической анемии. У них повышен риск образования онкопатологий.
Иммунологический анамнез
 Первым этапом специфической иммунологической диагностики является иммунологический анамнез. Это очень важная часть общего анамнеза больного, независимо от основного заболевания, позволяет выяснить наличие заболеваний в прошлом, установить или заподозрить иммунологическую причину заболевания или направление поиска иммунологических нарушений.В результате сбора анамнеза следует сразу выяснить тип вероятного иммунопатологического синдрома. Существуют различные точки зрения на количество и проявления иммунопатологических синдромов.На современном этапе, на наш взгляд, следует выделять 6 иммунопатологических синдромов. Эта точка зрения освещена также и Е.И.Соколовим (1998).
Первым этапом специфической иммунологической диагностики является иммунологический анамнез. Это очень важная часть общего анамнеза больного, независимо от основного заболевания, позволяет выяснить наличие заболеваний в прошлом, установить или заподозрить иммунологическую причину заболевания или направление поиска иммунологических нарушений.В результате сбора анамнеза следует сразу выяснить тип вероятного иммунопатологического синдрома. Существуют различные точки зрения на количество и проявления иммунопатологических синдромов.На современном этапе, на наш взгляд, следует выделять 6 иммунопатологических синдромов. Эта точка зрения освещена также и Е.И.Соколовим (1998).
Вторичный иммунодефицит
Под вторичным иммунодефицитом следует понимать наличие приобретенных болезней иммунной системы. В данном случае, как и при первичных иммунодефицитах, речь идет о слишком частых атаках инфекционных болезней в связи с ослабленным иммунитетом. Наиболее известным примером данного типа иммунодефицита является СПИД, который развивается в результате ВИЧ-инфекции. Кроме того, вторичные иммунодефициты проявляются под воздействием лекарственных средств, облучения, определенных хронических болезней. Вторичный иммунодефицит может наблюдаться у больных, которые обращаются к врачу с жалобами на самые разные недуги.
В целом, все действия, которые, так или иначе, вызывают ослабление иммунной системы человека, способствуют развитию у него вторичного иммунодефицита.
Кроме того, данное состояние возникает при пищевых дефицитах, при которых наблюдается белково-калорийная недостаточность, а также недостаток витаминов и микроэлементов. В данном случае особенно пагубно сказывается на состоянии человека дефицит витамина А, селена, цинка. Также в группу риска иммунодефицитов входят люди, имеющие хронические метаболические расстройства, возникающие вследствие болезней печени и почек. В некоторой степени подвержены развитию иммунодефицита и люди, перенесшие серьезную операцию либо травму.
При вторичных иммунодефицитах важно как можно как можно раньше обнаружить бактериальные инфекции и предпринять необходимое лечение
Вирус иммунодефицита человека
Вирус иммунодефицита человека принято относить к семейству ретровирусов. На сегодняшний день медики определяют два типа данного вируса — ВИЧ1 и ВИЧ2. Их принципиальные различия состоят в антигенных и структурных особенностях.
Вирус иммунодефицита человека не является устойчивым к влиянию особенностей внешней среды. Его уничтожает практически каждое вещество с дезинфицирующими свойствами. Считается, что этот вирус может быть в каждой биологической жидкости человеческого организма. Но при отсутствии крови в такой жидкости количества вируса недостаточно для того, чтобы произошло инфицирование. Следовательно, неопасными биологическими жидкостями считается слюна, пот, слезы, рвотная масса. В то же время в каждой жидкости, которая связана с лимфой, содержит вирус в больших количествах. Именно поэтому очень высок риск передачи ВИЧ при половых контактах, а также в процессе вскармливания ребенка грудью. Следовательно, самыми опасными жидкостями организма с точки зрения заражения ВИЧ являются кровь, вагинальные секреты, лимфа, сперма, цереброспинальная, асцитическая, перикардиальная жидкости, грудное молоко.
Вирус иммунодефицита человека, оказываясь в организме, попадает в клетки-мишени, которые являются регуляторами в процессе иммунного ответа. Постепенно вирус попадает в другие клетки, и патологический процесс происходит в разных системах и органах.
В процессе гибели клеток иммунной системы проявляется иммунодефицит, симптомы которого вызывает вирус. Под его действием у человека развиваются болезни, имеющие как инфекционный, так и неинфекционный характер.
Тяжесть болезни и скорость ее прогрессирования напрямую зависит от наличия инфекций, генетических особенностей организма человека, его возраста и др. Инкубационный период длиться от трех недель до трех месяцев.
После этого наступает стадия первичных проявлений, на которой у больного проявляются разнообразные клинические симптомы и активно продуцируются антитела. Эта стадия у разных людей может протекать по-разному. Возможно ее бессимптомное течение, наличие острой инфекции без вторичных заболеваний, а также инфекция с вторичными заболеваниями.





В процессе перехода вируса в субклиническую стадию постепенно нарастает иммунодефицит, лимфоузлы у человека увеличиваются, в то же время скорость размножения ВИЧ замедляется. Эта стадия достаточно длительная: она иногда длится до двадцати лет, хотя ее средняя продолжительность составляет около шести лет. Позже у больного развивается синдром приобретенного иммунодефицита.
Причины первичного иммунодефицита
Любое заболевание, поражающее иммунную систему, изучено не в полной мере. Также обстоят дела с ПИД. Однако причины данного заболевания все же выяснены.
Генетические мутации. В этом случае изменяются гены, способствующее образованию и развитию иммунокомпетентных клеток.
Тератогенное воздействие. В утробе матери развивается плод. Токсины различного происхождения угнетают его развития, вызывая изменения на генном уровне. TORCH-инфекции часто наблюдаются при тератогенном воздействии.
Неясная этиология. Под это понятие подходят те случаи, которые не удается выявить и точно обозначить.
Вообще, даже те причины, что конкретно обусловлены медицинской наукой, имеют еще ряд серьезных вопросов. Когда код ДНК будет полностью раскрыт, то наука сможет лечить болезни без ограничений. Тогда раскроются новые горизонты. Это важный момент, ведь перспектива медицинской науки уже заложена в настоящее время.
Первичные иммунодефициты классификация
Следует различать первичные и вторичные иммунодефициты. Первичный определяется у младенца вскоре после рождения. Его организм лишён возможности защищаться от антигенов, подвержен инфекционному вторжению. Это выражается в том, что малыш часто болеет, его одолевают повторные недуги, он трудно переносит их, получает осложнения. Тяжёлые формы первичного иммунодефицита приводят к смерти в младенчестве.
Известны редчайшие случаи, когда первичная иммунная недостаточность проявлялась у взрослых. Такое возможно, но для этого у человека должна быть высокая компенсированность определённой разновидности болезни.
Клиника заболевания — это повторное инфицирование, переход болезней в хроническую форму. К чему приводит первичный иммунодефицит:
- Пациент страдает бронхолегочными аномалиями.
- У него поражаются слизистые оболочки и кожный покров.
- Есть проблемы с ЛОР-органами.
- ПИДС, как правило, приводит к лимфадениту, абсцессам, остеомиелиту, менингиту, сепсису.
- Определённые формы первичного иммунодефицита провоцируют аллергии, аутоиммунные заболевания, рост злокачественных новообразований.
Изучением нарушений функций иммунной защиты занимается иммунология — наука о развитии и становлении защитного механизма, противодействующего проникновению антигенов в организм и уничтожающего повреждённые вредоносными веществами и микроорганизмами клетки.
Чем раньше диагностирован ПИДС, тем больше шансов у ребёнка выжить и продолжать жизнь с удовлетворительным состоянием здоровья
Важное значение имеет своевременное определение генной мутации, которая даёт возможность определиться с планированием семьи
Иммунодефицитом считается стойкая аномалия защитного механизма, которая порождает сбой в иммунном ответе на влияние антигенов. Этот сбой может быть четырёх видов:
- возрастной, то есть возникший в детстве, либо в преклонном возрасте;
- приобретённый из-за неправильного питания, образа жизни, приёма лекарств, вируса СПИД и т. д.;
- развившийся в результате различных инфекций;
- врождённый или первичный ИД.
ПИДС классифицируются в зависимости от форм и тяжести заболевания. К первичным иммунодефицитам относятся:
- ИД характеризующийся поражением нескольких клеточных комплексов;
- Дисгенез ретикулярный, при котором стволовые клетки отсутствуют, это обрекает новорождённого на смерть.
- Тяжёлый комбинированный ИД — наследственное заболевание, обусловленное дисфункцией В и Т-лимфоцитов.
- Синдром Ди Джорджа — или аномалии тимуса, паращитовидных желёз — недоразвитость, либо отсутствие вилочковой железы. В результате дефекта поражаются Т-лимфоциты, возникают врождённые пороки сердца, деформации в костной структуре, строении лицевых костей, почечные дефекты и дисфункции ЦНС.
- Первичный иммунодефицит, обусловленный поражением В-лимфоцитов.
- Нарушения в миелоидных клетках, провоцирующих хроническую гранулематозную болезнь (ХГБ) с аномалией в кислородном обмене. Дефект выработки активного кислорода влечёт хронические грибковые и бактериальные инфекции.
- Дефекты сложных белков крови, нарушающих гуморальную защиту. В системе комплемента может отсутствовать несколько составляющих.
Методы лечения
Основные методы лечения первичного иммунодефицита заключаются в осуществлении профилактики бактериальных, вирусных и грибковых инфекций, заместительной терапии ферментами, приёме иммуномодулирующих препаратов и витаминов.
Лекарственные препараты
Для поддержания функций пассивной иммунной системы используют следующие средства медикаментозной терапии:
- Мабтера — это инъекционный препарат на основе активного вещества ритуксимаб, который вводится в организм больного в составе внутривенных капельниц, является источником моноклональных антител (средняя стоимость медикамента 76000 руб. за флакон 500 мл);
- Пролиа — это терапевтический раствор для внутривенных инъекций, который усиливает защитные функции иммунной системы (цена препарата в аптеках 12900 руб. за флакон 60 мл);
- Альфарекин — это иммуномодулятор, который поддерживает ослабленную иммунную систему в борьбе с патогенными микроорганизмами (лекарственное средство предназначено для инъекционного введения, а его средняя стоимость составляет 7500 руб.);
- Циклоферон — это эффективный иммуномодулятор, который вводится в организм больного в виде внутримышечных или внутривенных инъекций (цена медикамента составляет 1800 руб. за 5 ампул по 2 мл).
Продолжительность терапии и дозировка вышеперечисленных препаратов определяется индивидуально в зависимости от разновидности первичного иммунодефицита, возраста пациента, наличия возможных осложнений.
Прочие методы
Одним из экспериментальных способов лечения первичного иммунодефицита является коррекция генетического дефекта с помощью генной инженерии, но эта терапевтическая методика ещё не совершенна.
В зависимости от типа патологического состояния иммунной системы может быть применено оперативное вмешательство, которое заключается в пересадке костного мозга от совместимого донора. Данный метод эффективен при лечении первичного иммунодефицита с нарушением клеточного звена иммунной системы. Операция проводится в стерильных условиях операционного зала под общей анестезией.




В отдельных случаях используют метод переливания нейтрофилов, а также замещение иммуноглобулинов. Все терапевтические манипуляции проводятся в клиниках и медицинских центрах, специализирующихся на лечении гематологических и генетических заболеваний крови.
В 2018 году отечественный препарат на основе иммуноглобулинов класса G успешно прошёл стадию клинических исследований. Медикамент полностью безопасен и может быть использован для комплексной терапии первичного иммунодефицита, но в массовое производство лекарственное средство пока ещё не поступило.
Клинические проявления
Для каждой нозологической формы иммунодефицита характерны свои симптомы и особенности течения. Однако существуют общие черты, характерные для всех первичных иммунодефицитов:
- Манифестация патологии иммунной системы с раннего возраста.
- Часто рецидивирующие хронические инфекционные заболевания ЛОР-органов, дыхательных путей, пищеварительной системы.
- Характерные возбудители этих болезней – условно-патогенная флора, грибы, простейшие.
- Склонность к генерализации инфекционного процесса и нечувствительность к традиционным методам лечения.
- Отставание прибавок роста и массы тела.
- Расстройства пищеварения (хроническая диарея и нарушение всасывания).
- Аутоиммунные процессы (артрит, СКВ-подобный синдром, тиреоидит).
- Аллергические реакции, в том числе на введение лекарственных препаратов и иммуноглобулинов.
- Необычные реакции на вакцинацию.
- Опухоли и лимфопролиферативные заболевания.
- Изменение в анализе крови (анемия, снижение концентрации лейкоцитов и тромбоцитов).
- Сочетание с пороками развития (гипоплазия клеточных элементов хряща и волос).
Согласно международным рекомендациям выделены признаки, наличие которых должно насторожить врача в отношении первичного иммунодефицита, к ним относятся:
- частые отиты и синуситы (6 и более раз в год);
- повторные пневмонии (2 раза в год и чаще);
- неоднократно перенесенный сепсис, менингит или остеомиелит;
- дефицит массы у ребенка в сравнении с возрастной нормой и отставание в росте;
- грибковые поражения кожи и слизистых у ребенка после года;
- гнойничковая сыпь на коже и абсцессы внутренних органов;
- лимфоаденопатия и увеличение печени;
- частое использование антибиотиков для купирования инфекционных процессов;
- наличие врожденных иммунодефицитов у родственников и случаев ранней смерти детей от инфекционных заболеваний.
Выявление хотя бы одного из этих признаков является показанием для углубленного обследования иммунной системы у ребенка.
Первичные дефициты белков комплемента
Недостаточность белков комплемента проявляется по-разному в зависимости от того, какой (или какие) белки отсутствуют.
Выделяют три группы заболеваний, связанных с первичным дефицитом комплемента:
- Комплемент-зависимые иммунодефицитные синдромы
- Комплемент-ассоциированные аутоиммунные болезни
- Наследственный ангионевротический отёк Квинке—Ослера.
Комплемент-зависимые иммунодефицитные синдромы
Комплемент-зависимые иммунодефицитные синдромы — заболевания, сопровождающиеся недостаточностью антибактериальной защиты организма. Они проявляются частыми инфекционными процессами в различных органах и тканях. Поскольку белки комплемента при активации играют роль хемоаттрактантов и опсонинов, обеспечивая эффективную функцию фагоцитирующих клеток, то при дефиците компонентов комплемента формируется вторичная недостаточность функции макрофагов и нейтрофильных гранулоцитов. Особенно часто инфекционные процессы при этом вызваны стрептококками, в частности пневмококками, и Haemophilus influenzae. В эту группу включают недостаточность С3b-инактиватора, белков С3, С6 и С8.
Недостаточность С3b-инактиватора. С3b-инактиватор играет роль ингибитора альтернативного пути активации комплемента. При его отсутствии происходит быстрое потребление С3-компонента (вторичный дефицит С3), который в нормальных условиях принимает активное участие в антибактериальной защите организма. Белка С3 у больных в плазме примерно 20 % от нормы. Однако на 75 % он представлен С3b-фрагментом. Уровень нативного С3 составляет всего 5 % от нормы. Скорость расщепления С3 у больных повышена почти в 5 раз. Показано, что через 2 часа после инъекции нативного С3 расщеплению подвергается 40 % введённых молекул. Помимо вторичного дефицита С3 формируется вторичная недостаточность белка С5, однако она менее выражена (примерно 40 % от нормального уровня). Заметно снижена концентрация фактора В — 5 % от нормы (расщепление фактора В происходит под влиянием фактора D). Уровень пропердина снижен незначительно. Больные при этом заболевании страдают различными бактериальными инфекциями.
Недостаточность С3. Недостаточность С3-компонента комплемента также проявляется различными бактериозами. В основе заболевания, в отличие от недостаточности С3b-инактиватора, лежит первичный дефицит С3-белка.
Комплемент-ассоциированные аутоиммунные болезни
Недостаточность белков комплемента провоцирует возникновение аутоиммунных заболеваний, прежде всего (1) красной волчанки, (2) так называемого волчаночно-подобного синдрома и (3) ревматоидного артрита. Часто поражаются почки по типу гломерулонефрита. У больных также описаны пурпура Шёнлейна—Геноха и полимиозит. К этим заболеваниям относятся недостаточность белков С1, С2, С4 и С5. Гены этих белков сцеплены с генами иммунного ответа (генами МНС), поэтому дефекты их, как правило, обоюдны.
Недостаточность С2. Недостаточность С2 является самым частым вариантом первичного дефицита белков комплемента. С2 синтезируют фиксированные и блуждающие макрофаги, фагоцитарная функция которых при этом не нарушена.
Наследственный ангионевротический отёк Квинке—Ослера
К третьей группе состояний, связанных с первичной недостаточностью комплемента, относится наследственный ангионевротический отёк Кви́нке—О́слера, в основе которого лежит недостаточность С1-ингибитора. У отдельных больных при этом возникают аутоиммунные процессы, прежде всего красная волчанка.