Диагностика
На сегодняшний день генная мутация идентифицирована и
существуют специальные программы диагностического тестирования, разработанные
исследовательским фондом прогерии.
В данный момент имеется возможность подтвердить определенные
мутации в гене или генетические изменения, приводящие к СХГП. К тому же, наука
на месте не стоит и сейчас учеными разрабатывается окончательный научный
метод диагностики детей.
Естественно, это поспособствует ранней и более точной
диагностике. Пока же в медицинских заведениях ребенка с таким диагнозом
осматривают внешне, берут анализы и для тестирования — образец крови.
Прогерия симптомы
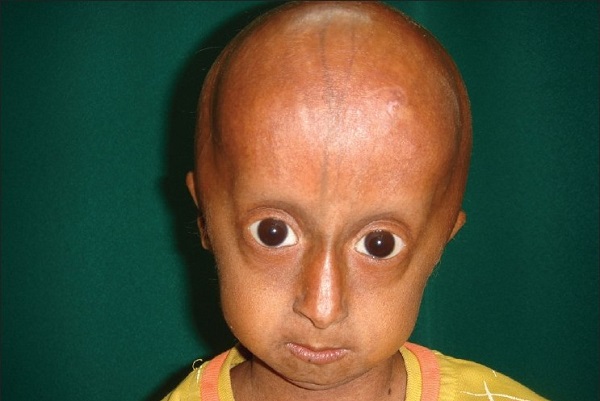
Клиническая картина детской прогерии отличается характерным преждевременным атеросклерозом, фиброзом миокарда, нарушениями мозгового кровообращения, повышением липопротеидов и уровнем холестерина, протромбиновым временем в анализах, ранними инфарктами, скелетными аномалиями. В данном случае имеются выраженные диспропорции лица и черепа, недоразвитие челюсти и зубов, смещение бёдер. Длинные кости при нормальной корковой структуре и прогрессировании периферической деминерализации подвергаются рецидивирующим патологическим переломам.
Суставам характерна тугая подвижность, особенно коленным с возможными контрактурами тазобедренных, голеностопных, локтевых и лучезапястных суставов. При рентгенологических исследованиях обнаруживается деминерализация около суставов с остеопорозом, варусными и вальгусными деформациями нижних конечностей. Также очень часто развиваются опухоли и утолщение коллагеновых волокон.
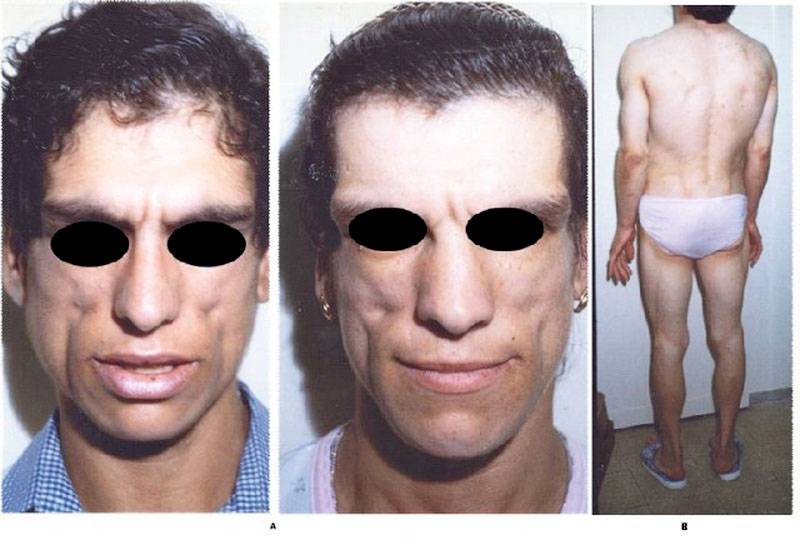
Синдром Вернера или взрослая прогерия проявляется от 14 до 18 лет и характеризуется отставанием в росте, универсальным поседением с параллельным прогрессированием алопеции.
Как правило, синдром прогерия развивается после двадцати лет и отличается ранним облысением, истончением кожных покровов на лице и конечностях, характерной бледностью. Под слишком натянутой кожей просматриваются поверхностные кровеносные сосуды, а подкожная жировая клетчатка и мышцы, расположенные под ней, полностью атрофируются, поэтому конечности выглядят непропорционально тонко.
Затем кожа над выступами костей постепенно становится толще и изъязвляется. После тридцати лет у больных прогерией развивается катаракта обоих глаз, голос становится слабым, высоким и хриплым, заметно поражаются кожные покровы. Это проявляется в виде склероцермоподобных изменений конечностей и лица, сухости кожи, язв на ногах, мозолей на стопах и телеангиэктазией. Такие больные, как правило, низкого роста, с лунообразным лицом, клювоподобным носом, как у птицы, суженным ротовым отверстием и выступающим резко подбородком, полным туловищем и тонкими конечностями.
У больных прогерией нарушаются функции потовых и сальных желёз. На выступах костей образовывается гиперкератоз, проявляется общая гиперпигментация, изменяется форма ногтевых пластин. А после различных травм на голенях и стопах появляются трофические язвы. Кроме атрофии и истончений, у больных отмечаются значительные изменения в мышцах и костях, кальцификация, остеопороз генерализованного характера, остеоартриты с эрозиями. Такие больные ограничены в движениях пальцев и сгибательной контрактуре. Для больных прогерией свойственна деформация костей, как при ревматоидном артрите, боль в конечностях, плоскостопие и остеомиелит.
Во время обследований на рентенографии выявляются остеопороз костей, гетеротопические кальцинаты кожи и подкожной клетчатки, связок и сухожилий. Также, медленно прогрессирует катаракта, развивается атеросклероз, нарушающий деятельность сердечно-сосудистой системы. У большинства больных снижается интеллект.
После сорока лет к прогерии на фоне сахарного диабета, дисфункций паращитовидных желёз и других заболеваний почти у 10% пациентов развиваются опухолевые патологии в виде остеогенной саркомы, астроцитомы, тиреоидной аденокарциномы, рака молочной железы и кожи.
Летальный исход обычно является следствием сердечно-сосудистых патологий и злокачественных опухолей.
При гистологическом анализе синдрома прогерии устанавливают атрофию придатков кожи, где сохраняются эккринные железы; дерма при этом имеет утолщение, гиалинизируются волокна из коллагена, а нервные волокна разрушаются.
У больных полностью атрофируются мышцы, отсутствует подкожный жир.
Заболевание диагностируется на основании клинической симптоматики прогерии. При сомнениях в диагнозе определяют способность фибробластов размножаться в культуре (сниженный показатель для синдрома Вернера). Для дифференциального диагноза прогерии учитывают синдромы Хатчинсона-Гилфорда, Ротмунда-Томсона и системную склеродермию.
Детская прогерия Хатчинсона-Гилфорда
Это болезнь встречается очень изредка в соотношении 1:4000000 новорождённых в Нидерландах и 1:8000000 в США. Причём болезнь поражает больше мальчишек, чем девченок (1,2:1).
Рассматривают две формы прогерии Хатчинсона-Гилфорда: традиционную и неклассическую.
В истинное время описано более 100 случаев детской прогерии. Причём в главном это болезнь поражает малышей белоснежной расы. Для прогерии Хатчинсона-Гилфорда характерно полиморфное поражение. Детки, имеющие таковой синдром, смотрятся полностью нормальными при рождении. Но уже к году либо двум наблюдается серьёзное отставание в росте. Как правило такие детки отличаются очень небольшим ростом и ещё более низкой массой тела в согласовании с его длиной.
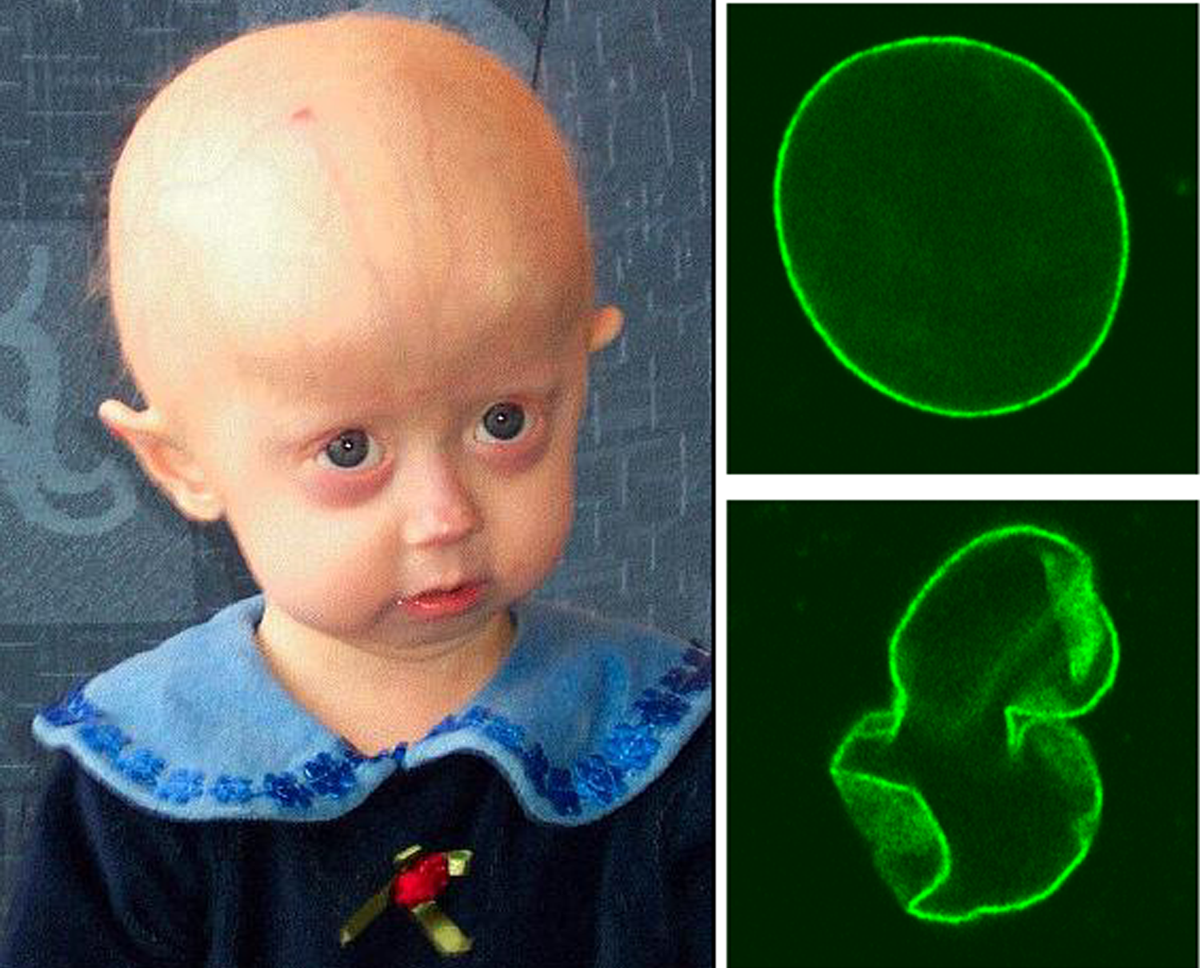






Для малышей с прогерией типично полное облысение не только лишь волосистой части головы, да и отсутствие ресниц, бровей ещё с ранешнего возраста. Кожа смотрится слабенькой и морщинистой в итоге абсолютной утраты подкожного жира, находится цианоз кожных покровов. Для головы свойственна несимметричность черепно-лицевых костей, которые напоминают лицо птицы с крючковатым носом, аномально маленький нижней челюстью, выпученными глазными яблоками и оттопыренными ушами. Конкретно эти черты, большая плешина и малая челюсть, присваивают наружности ребёнка вид старенького человека.
Другие клинические проявления прогерии включают: неверное и позже прорезывание зубов, узкий и высочайший глас, грушевидная грудная клеточка и уменьшенные в размерах ключицы. Конечности как правило тонкие, а изменённые локтевые и коленные суставы присваивают нездоровому ребёнку «позу наездника».
У малышей ещё до года отмечаются склероподобные уплотнения, врождённого либо приобретённого нрава, на ягодицах, бёдрах и понизу животика. Детям с прогерией свойственна гиперпигментация кожи, которая только усиливается с возрастом и гипоплазия ногтей, при которой они становятся жёлтыми, тонкими и выпуклыми, напоминающие часовые стёкла. Но, начиная с пятилетнего возраста, развивается распространённая форма атеросклероза с огромным поражением аорты и артерий, в особенности брыжеечных и коронарных. А уже еще позднее возникают сердечные шумы и гипертрофия сердца, в левом желудочке. Преждевременное появление у малышей атеросклероза, становится предпосылкой непродолжительности их жизни. А вот основной предпосылкой погибели считается инфаркт миокарда.
При прогерии известны случаи ишемического инфаркта. Такие детки в интеллектуальном развитии полностью ничем не отличаются от здоровых малышей, время от времени даже опережают их. Детки с таким диагнозом в среднем живут около 14-ти лет.
При детской прогерии неклассической формы длина тела от массы отстаёт некординально, в протяжении долгого времени волосы сохраняются, а липодистрофия прогрессирует еще медлительнее; вероятен рецессивный тип наследования.
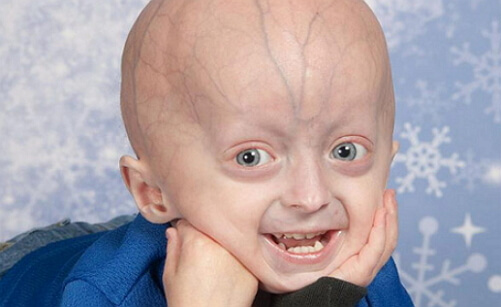
детская прогерия фото
Фатальная семейная бессонница
www.libertatea.ro
Фатальная семейная бессонница — редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в сорока семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того как заболевание прогрессирует, пациент страдает от все более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает — как правило, от истощения или пневмонии.
Смертельная бессонница возникает из-за того, что в кодоне (кодирующем тринуклеотиде) 178 гена PRNP, расположенного в 20-й хромосоме, вместо аспарагина, который в норме присутствует в организме в составе белков и необходим для нормальной работы нервной системы, появляется аспарагиновая кислота. Это меняет форму белковой молекулы, и она превращается в прион — агрессивный аномальный белок, в составе которого нет нуклеиновых кислот. Под действием одного приона окружающие молекулы уподобляются ему, и это ведет к необратимым изменениям. Прежде чего, они протекают в тканях таламуса: подкорковой «станции» всех видов чувствительности, отвечающей в том числе за возникновение сонного состояния и двигательные функции: глотание, сосание, жевание, смех. Под действием прионов ядра таламуса покрываются порами, превращаются в губку и перестают работать.
Для болезни характерен аутосомно-доминантный тип наследования: то есть у нее нет носителей. Детям она передается от родителей с вероятностью 50% и только при условии, если кто-то из них болен. Мужчины и женщины болеют фатальной семейной бессонницей с одинаковой частотой. На сегодняшний день это заболевание считается неизлечимым.
СИНДРОМ ХАТЧИНСОНА-ГИЛФОРДА ИЛИ ДЕТСКАЯ ПРОГЕРИЯ
- Подробности
- Опубликовано: 05 декабря 2018
- Просмотров: 402
Vasilchenko T.S., Gabdrakipova A.A.
Vasilchenko Tatiana Sergeevna – Student;
Gabdrakipova Albina Aybekovna — Student,
DEPARTMENT OF FACULTY THERAPY, FACULTY MEDICAL TREATMENT,
MEDICAL INSTITUTE
BELGOROD STATE NATIONAL RESEARCH UNIVERSITY,
BELGOROD
Abstract: this article studies a very rare genetically determined disease — Hutchinson-Gilford Syndrome or childhood progeria. The main clinical forms of this disease are considered. The causes and molecular mechanisms of childhood progeria are analyzed. The main clinical manifestations of premature aging are described. It is noted that this syndrome affects all tissues, organs and systems of the body. The main causes of death of patients with this disease, such as myocardial infarction caused by generalized atherosclerosis of large vessels, and malignant neoplasms are distinguished.





Keywords: progeria, premature aging, Hutchinson-Gilford syndrome, genetic diseases.
Васильченко Т.С., Габдракипова А.А.
Васильченко Татьяна Сергеевна – студент;
Габдракипова Альбина Айбековна – студент,
кафедра факультетской терапии, факультет лечебного дела,
Медицинский институт
Белгородский государственный национальный исследовательский университет,
г. Белгород
Аннотация: в данной статье изучается очень редкое генетически обусловленное заболевание — Синдром Хатчинсона-Гилфорда или детская прогерия. Рассматриваются основные клинические формы данного заболевания. Анализируются причины и молекулярные механизмы возникновения детских прогерий. Описываются основные клинические проявления преждевременного старения. Отмечается, что при данном синдроме поражаются все ткани, органы и системы организма. Выделяются основные причины смерти больных этим заболеванием, такие как инфаркт миокарда, обусловленный генерализованным атеросклерозом крупных сосудов, и злокачественные новообразования.
Ключевые слова: прогерия, преждевременное старение, синдром Хатчинсона-Гилфорда, генетические заболевания.
Список литературы / References
- Makhotin Y.V., Karev O.V., Losev T.N. A book about health:sourcebook. M.: Meditsina, 1988. 417 p. Russian (Махотин Ю.В., Карева О.В., Лосева Т.Н. Книга о здоровье. М.: Медицина, 1988. 417 с.).
- Gilford H. On a condition of mixed premature and immature development. Medico-Chirurgical Transactions, 1897 (80):17–45.
- Gilford H. Progeria: a form of senilism. Practitioner, 1904 (73): 188–217.
- Skripkina Yu.K. ed. Skin and venereal diseases. M.: Meditsina, 1996. 305 p. Russian (Кожные и венерические болезни / под ред. Ю.К. Скрипкина. М.: Медицина, 1996. 305 с.).
- Burtner R.C., Kennedy B.K. Progeria syndromes and ageing: what is the connection? Nature review. Molecular cell biology, 2010. 11: 567–578.
- Fedorov E.V. et al. About congenital progeria. Pediatrics, 1980 (4): 66. Russian (Федорова Е.В. и др. О врожденной прогерии. Педиатрия, 1980 (4): 66).
- Козлова С.И., Демикова Н.С., Семанова Е. и др. Наследственные синдромы и медико-генетическое консультирование. М.: Практика, 1996. 230 с.
Ссылка для цитирования данной статьи
 |
Тип лицензии на данную статью – CC BY 4.0. Это значит, что Вы можете свободно цитировать данную статью на любом носителе и в любом формате при указании авторства. | |
|
Ссылка для цитирования. Васильченко Т.С., Габдракипова А.А. СИНДРОМ ХАТЧИНСОНА-ГИЛФОРДА ИЛИ ДЕТСКАЯ ПРОГЕРИЯ // VIII INTERNATIONAL SCIENTIFIC REVIEW OF THE PROBLEMS OF NATURAL SCIENCES AND MEDICINEСвободное цитирование при указании авторства: https://scientific-conference.com/grafik/2018-vtoroe-polugodie.html(Boston, USA — 04 December, 2018). с. {см. сборник} |

Коллагеновая недостаточность
С возрастом в коже снижается синтез коллагена, она становится менее упругой, появляются морщины и обвисание. Мутации разных генов могут приводить к нарушению синтеза коллагена с самого рождения — эти состояния встречаются даже у животных и могут проявляться по-разному. С недостаточностью разных типов коллагена связаны, например, несовершенный остеогенез и синдром Элерса — Данлоса. Если страдает именно коллаген кожи, то проявления коснутся в первую очередь внешнего вида. Синдром вялой кожи (он же эластолиз, или cutis laxa на латыни) встречается примерно у одного ребёнка из двух миллионов. По данным ассоциации Cutis Laxa Internationale, в мире известно 385 пациентов с этим синдромом — впрочем, учитывая, что карта на сайте не отражает ни одного случая в России, на самом деле их больше.
Выделяют много разных видов эластолиза, вызванных генетическими мутациями, и приобретённую форму, причины которой неизвестны. Низкая эластичность кожи может быть изолированной, а может сопровождаться проблемами со связками, суставами, сердцем или сосудами. Пластический хирург, доктор медицинских наук, профессор Александр Тепляшин рассказывает, что точный диагноз не всегда можно установить — и поскольку речь о редких синдромах, то бывают случаи, когда приходит человек с уникальным набором проявлений. Также нужно понимать, что эффект эстетических операций не всегда достаточно выражен или устойчив: ткани с недостаточностью коллагена плохо удерживаются на месте. Чтобы остановить процесс, приведший к cutis laxa, или обратить его вспять, нужны новые виды терапии; для этого начинают применять, например, стволовые клетки, но пока в экспериментальном порядке.
Журналистка Виктория Аскеро-Дубовик рассказала, что окончательный диагноз ей не поставил ни один врач. По результатам множества обследований выяснилось, что все системы органов работают в соответствии с возрастом, а кожа будто опережает остальной организм на десятки лет: «Изменения в своей внешности я начала замечать в школе, кожа лица и тела выглядела намного старше моих лет. На лбу и шее были глубокие морщины, кожа обвисала. Но приёмы у докторов проходили как у обычно, результаты анализов соответствовали и соответствуют возрасту, а внешний вид врачей не беспокоил.
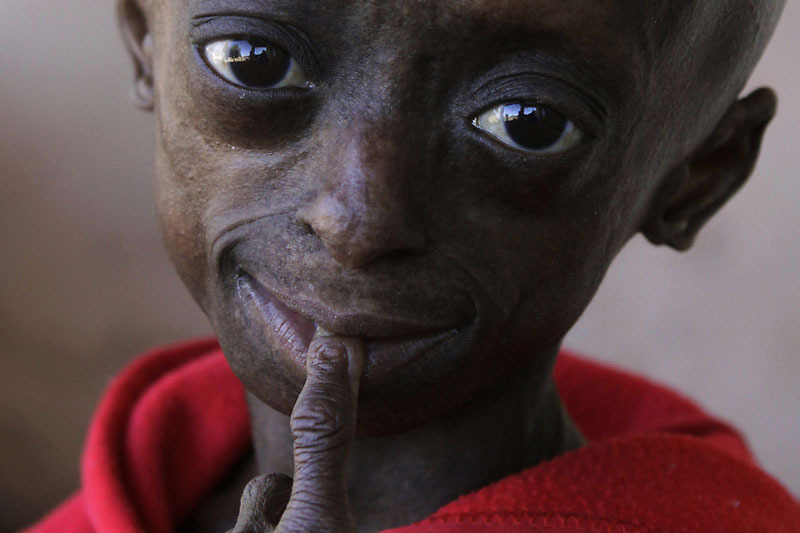
Прогерия симптомы
Клиническая картина детской прогерии отличается соответствующим ранним атеросклерозом, фиброзом миокарда, нарушениями мозгового кровообращения, увеличением липопротеидов и уровнем холестерина, протромбиновым временем в анализах, ранешними инфарктами, скелетными аномалиями. В этом случае имеются выраженные диспропорции лица и черепа, недоразвитие челюсти и зубов, смещение бёдер. Длинноватые кости при обычной корковой структуре и прогрессировании периферической деминерализации подвергаются рецидивирующим патологическим переломам.
Суставам свойственна тугая подвижность, в особенности коленным с вероятными контрактурами тазобедренных, голеностопных, локтевых и лучезапястных суставов. При рентгенологических исследовательских работах находится деминерализация около суставов с остеопорозом, варусными и вальгусными деформациями нижних конечностей. Также очень нередко развиваются опухоли и утолщение коллагеновых волокон.
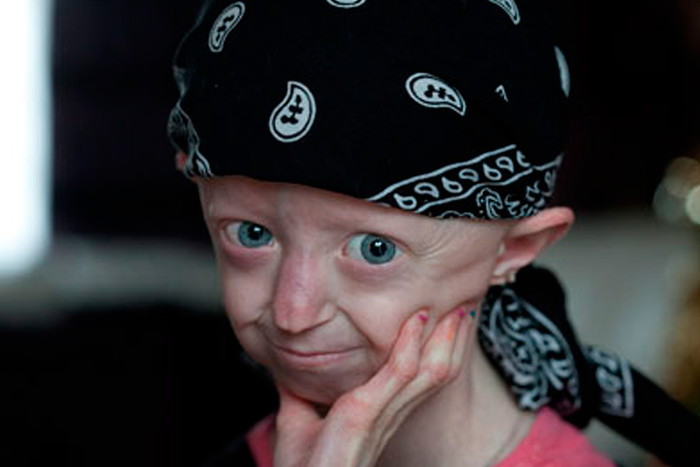






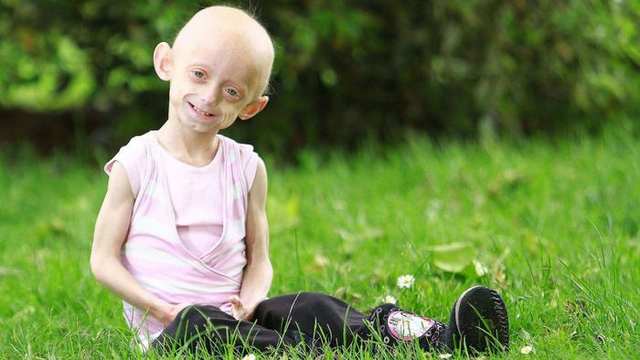
Синдром Вернера либо взрослая прогерия проявляется от 14 до 18 лет и характеризуется отставанием в росте, универсальным седением с параллельным прогрессированием алопеции.
Как правило, синдром прогерия развивается после 20 лет и отличается ранешным облысением, истончением кожных покровов на лице и конечностях, соответствующей бледностью. Под очень натянутой кожей просматриваются поверхностные кровяные сосуды, а подкожная жировая клетчатка и мускулы, расположенные под ней, на сто процентов атрофируются, потому конечности смотрятся диспропорционально тонко.
Затем кожа над выступами костей равномерно становится толще и изъязвляется. После 30 лет у нездоровых прогерией развивается катаракта обоих глаз, глас становится слабеньким, высочайшим и осиплым, приметно поражаются кожные покровы. Это проявляется в виде склероцермоподобных конфигураций конечностей и лица, сухости кожи, язв на ногах, мозолей на стопах и телеангиэктазией. Такие нездоровые, обычно, низкого роста, с лунообразным лицом, клювоподобным носом, насколько у птицы, суженным ротовым отверстием и выступающим резко подбородком, полным туловищем и тонкими конечностями.
У нездоровых прогерией нарушаются функции потовых и сальных желёз. На выступах костей создается гиперкератоз, проявляется общая гиперпигментация, меняется форма ногтевых пластинок. А после разных травм на голенях и стопах возникают трофические язвы. Не считая атрофии и истончений, у нездоровых отмечаются значимые конфигурации в мышцах и костях, кальцификация, остеопороз генерализованного нрава, остеоартриты с эрозиями. Такие нездоровые ограничены в движениях пальцев и сгибательной контрактуре. Для нездоровых прогерией характерна деформация костей, насколько при ревматоидном артрите, боль в конечностях, плоскостопие и остеомиелит.
Во время обследований на рентенографии выявляются остеопороз костей, гетеротопические кальцинаты кожи и подкожной клетчатки, связок и сухожилий. Также, медлительно прогрессирует катаракта, развивается склероз, нарушающий деятельность сердечно-сосудистой системы. Практически у всех нездоровых понижается ум.
После сорока лет к прогерии на фоне сладкого диабета, дисфункций паращитовидных желёз и других болезней практически у 10% пациентов развиваются опухолевые патологии в виде остеогенной саркомы, астроцитомы, тиреоидной аденокарциномы, рака молочной железы и кожи.
Летальный финал как правило является следствием сердечно-сосудистых патологий и злокачественных опухолей.
При гистологическом анализе синдрома прогерии устанавливают атрофию придатков кожи, где сохраняются эккринные железы; дерма при всем этом имеет утолщение, гиалинизируются волокна из коллагена, а нервные волокна разрушаются.
У нездоровых стопроцентно атрофируются мускулы, отсутствует подкожный жир.
Заболевание диагностируется на основании медицинской симптоматики прогерии. При колебаниях в диагнозе определяют способность фибробластов плодиться в культуре (сниженный показатель для синдрома Вернера). Для дифференциального диагноза прогерии учитывают синдромы Хатчинсона-Гилфорда, Ротмунда-Томсона и системную склеродермию.
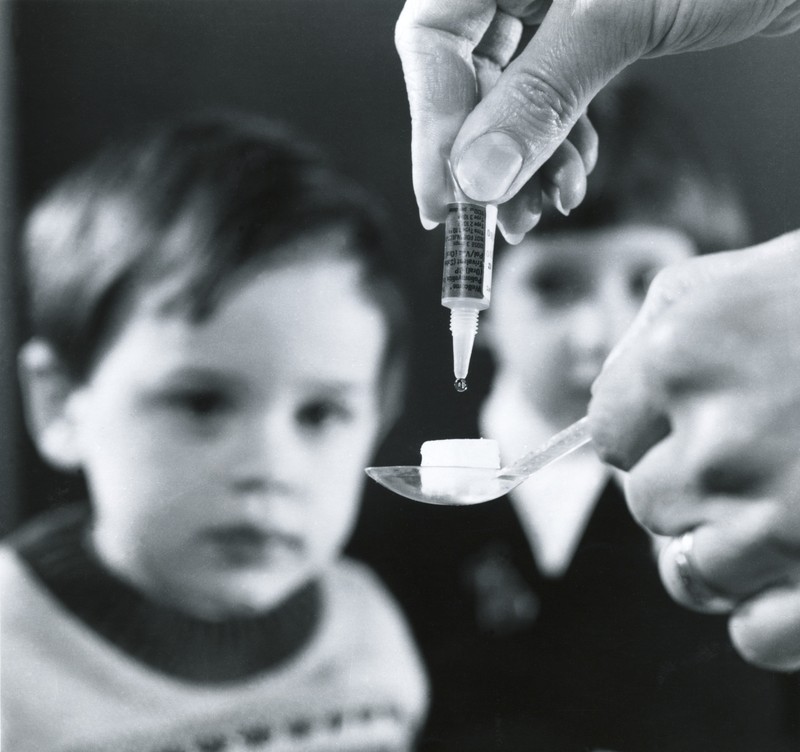
Прогерия лечение
До реального времени не существует определенного исцеления прогерии, его ещё не разработали. В главном терапия носит симптоматический нрав с профилактикой осложнений после атеросклероза и в исцелении трофических язв, сладкого диабета.
Для анаболического эффекта назначается СТГ, который у неких нездоровых наращивает массу тела и длину. Весь терапевтический процесс проводится рядом профессионалов, таких насколько эндокринолог, терапевт, кардиолог, онколог и других, зависимо от превалирующей симптоматики.
Но в 2006 году исследователями США было отмечено прогрессирование в лечении прогерии, насколько неизлечимого заболевания. Они занесли в культуру нарушенных фибробластов ингибитор фарнезилтрансферазы, который ранее проходил тесты на онкологических нездоровых. И этот процесс возвратил стареющим клеточкам нормальную форму. Таковой продукт был отлично перенесён, потому на данный момент существует надежда, что в дальнейшем появится возможность в его применении, чтоб предупредить прогерию ещё в детском возрасте.
Эффективность Лонафарниба (ингибитора фарнезилтрансферазы) заключается в увеличении количества жира под кожей, в массе тела, минерализации костей, что в конечном итоге уменьшит переломы.
Но, все же, пока это болезнь характеризуется неблагоприятными прогнозами. В среднем нездоровые прогерией доживают до тринадцатилетнего возраста, умирая от кровоизлияний и инфарктов.
Warning: date() [function.date
]: It is not safe to rely on the system’s timezone settings. You are *required* to use the date.timezone setting or the date_default_timezone_set() function. In case you used any of those methods and you are still getting this warning, you most likely misspelled the timezone identifier. We selected ‘Europe/Moscow’ for ‘MSK/3.0/no DST’ instead in /home/k45201/public_html/angina03.ru/mycode/main.php on line 3
15.04.2019
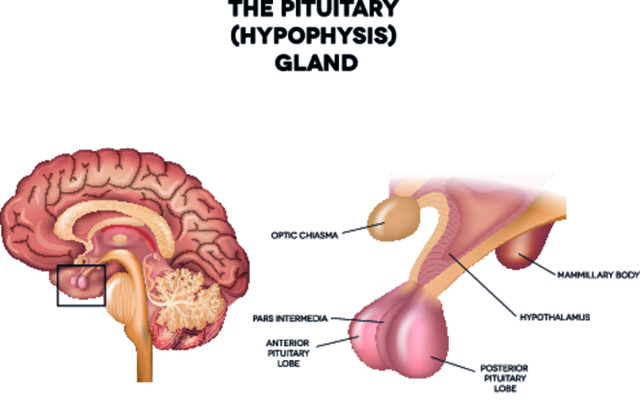





Синдром РОХХАД
www.cbsnews.com
Синдромом РОХХАД (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) — чрезвычайно редкое заболевание, при котором человек начинает стремительно набирать вес и страдает от булимии, респираторных болезней, остановок дыхания во сне, альвеолярной гиповентиляции и кардиореспираторных остановок. Также для пациентов с таким диагнозом характерно отсутствие реакции на повышение в крови углекислого газа.
На сегодняшний день в мире зарегистрировано порядка 100 случаев возникновения этого расстройства. Обычно оно проявляется в возрасте до 10 лет (чаще всего около 3 лет) и, по всей видимости, имеет наследственную природу. Несмотря на проведенные на Западе исследования, этиология синдрома РОХХАД до сих пор не ясна. Считается, что он появляется из-за дисфункции гипофиза, которую вызывает генетическая мутация. Однако ученым еще только предстоит определить, какой именно процесс нарушается в этом случае.
Вторая гипотеза развития синдрома Гетчинсона — Гилфорда
По другой теории, у больных прогерией снижено содержание антиоксидантов в организме, что приводит к ускорению перекисного окисления липидов, увеличению числа свободных радикалов, повреждению мембранного бислоя клетки и нарушению работы мембранных белков.
Закономерностей в возникновении детской прогерии не выявлено по причине малого числа зафиксированных случаев (не более 100). Известно, что в нескольких семьях синдром Гетчинсона — Гилфорда зарегистрирован у сибсов (потомки одних родителей), в том числе и от кровнородственных браков. В отношении типа наследования данного заболевания существуют различные точки зрения.
Симптоматика
Детская прогерия симптомы имеет следующие:
- маленький рост;
- отсутствие подкожной клетчатки;
- расширенная вена под кожей;
- несоразмерно большой череп;
- отсутствие волос на голове;
- плохое физическое развитие;
- большие глаза;
- дефекты зубов;
- «килевидная грудь»;
- высокий голос.
Несмотря на отставание в физическом развитии, дети с синдромом Хатчинсона-Гилфорда интеллектуально развиты, не отстают от сверстников в психическом развитии. Детская прогерия сопровождается прогрессированием атеросклероза уже с 5 лет и нарастанием сердечной патологии — появляются шумы при аускультации, симптомах гипертрофии миокарда. Кардиологические заболевания – самая частая причина смерти.
Случаи прогерии у взрослых, то есть синдром Вернера, характеризуются следующими состояниями:
- ранняя седина и облысение;
- появление старческих морщин в молодом возрасте;
- пигментация, сухость кожи;
- фиброзные уплотнения в подкожной клетчатке;
- голос становится глухим.
Прогерия – причина бесплодия мужчин и женщин. На поздних стадиях заболевания появляются трофические язвы на голенях. Из-за мышечной атрофии истончаются конечности, развиваются контрактуры суставов, плоскостопие. Характерна «поза всадника» из-за полусогнутых рук. Деформируются кисти, ногти желтеют, приобретают вид «часовых стекол».
При рентгенографии наблюдается остеопороз и отложение извести в околосуставных тканях, связочном аппарате суставов. Прогерию взрослых часто сопровождают доброкачественные опухоли различной локализации, эндокринные заболевания, сахарный диабет. В 8-12% возникают злокачественные опухоли. Поэтому прогерия симптомы имеет часто смазанные.
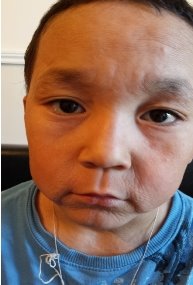
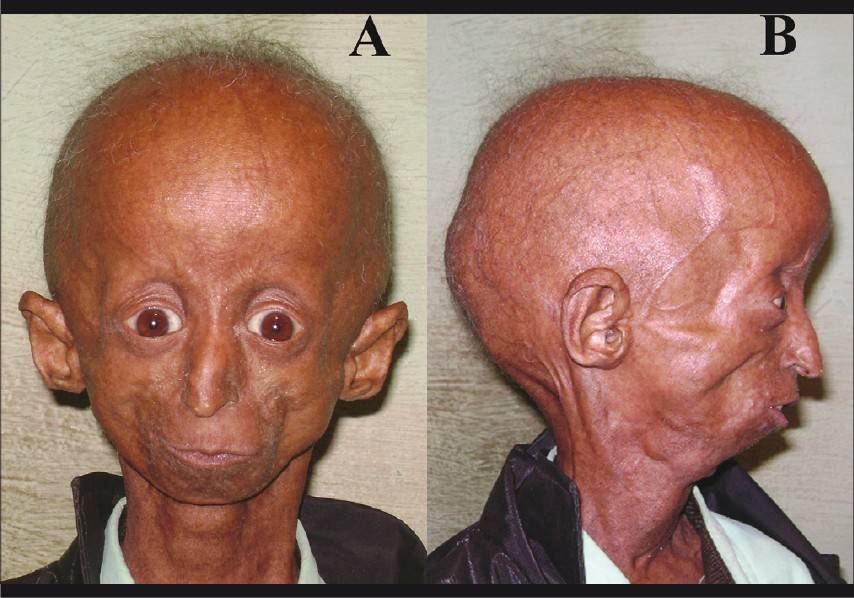
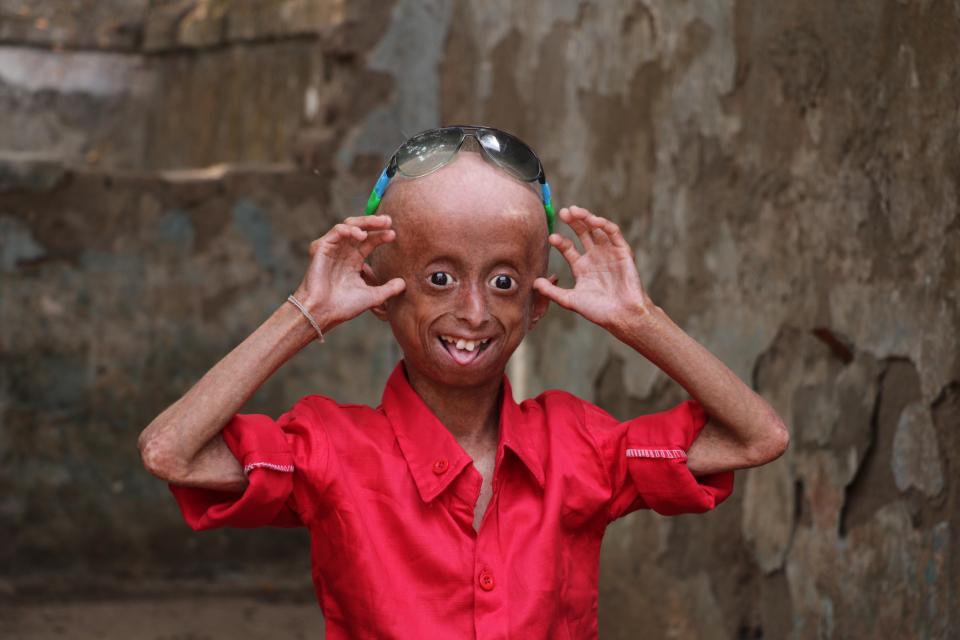
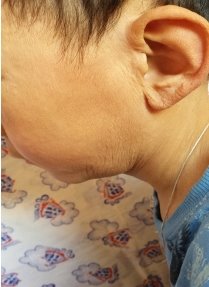
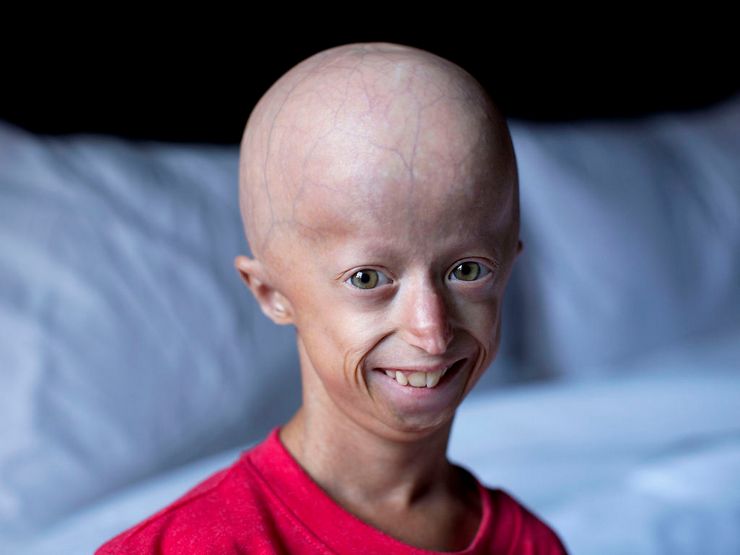
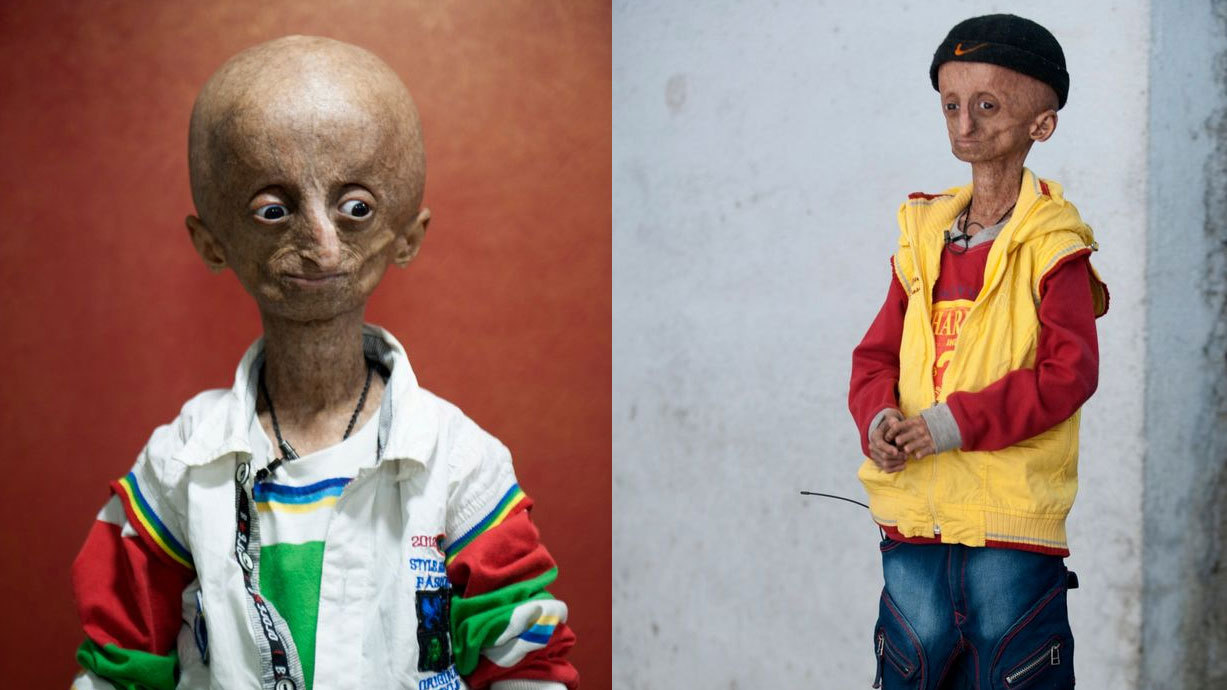
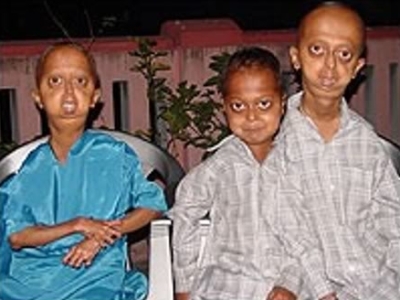
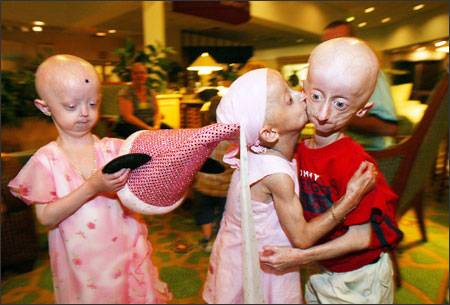
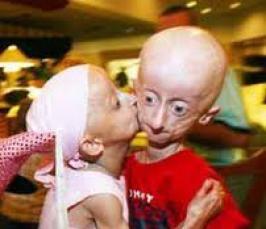
 Фото детей, страдающих прогерией
Фото детей, страдающих прогерией
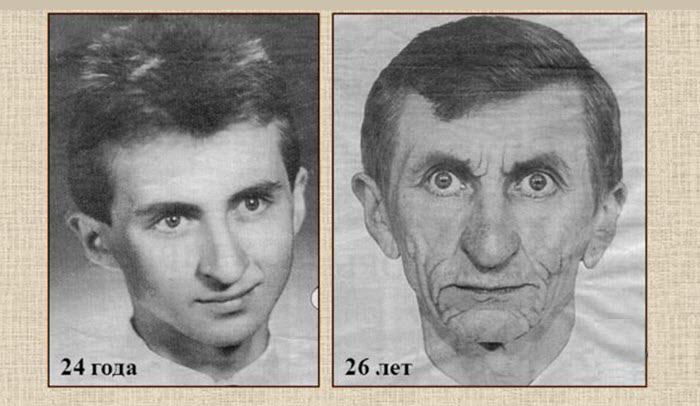
Похищенная молодость
Случаи внезапного старения весьма прозаичны: живущий в нормальных условиях ребенок поначалу удивляет окружающих своим быстрым развитием. В малолетнем возрасте он выглядит как совершеннолетний, а затем у него начинают проявляться все признаки… приближающейся старости.

В 1716 году в английском городе Ноттингеме умер восемнадцатилетний сын графа Уильяма Шеффилда, начавший стареть в тринадцатилетнем возрасте. Молодой Шеффилд выглядел намного старше своего отца: cедые волосы, наполовину выпавшие зубы, морщинистая кожа. У злосчастного юноши был вид потрепанного жизнью мужчины, он очень от этого страдал и принял смерть как избавление от мук.
Есть случаи подобного рода и среди представителей королевских родов. Венгерский король Людвиг II в девятилетнем возрасте уже достиг полового созревания и с удовольствием развлекался с придворными девицами. В четырнадцать он обзавелся густой окладистой бородой и стал выглядеть минимум на 35 лет. Год спустя он женился, а к шестнадцатилетию супруга подарила ему сына. Но в восемнадцать лет Людвиг полностью поседел, а еще два года спустя скончался со всеми признаками старческого одряхления.
Любопытно, что ни сын короля, ни дальнейшие его потомки подобной болезни не унаследовали. Из примеров ХIХ века можно выделить историю простой деревенской девушки, француженки Луизы Равальяк. В восьмилетнем возрасте Луиза, полностью сформировавшаяся как женщина, забеременела от местного пастуха и родила вполне здорового ребенка. К шестнадцатилетию у нее уже было трое детей и она выглядела старше своей матери, в 25 она превратилась в дряхлую старуху и, не дожив до 26, умерла от старости.
Не меньший интерес вызывают судьбы тех, кто жил в XX веке. Кое-кому из них повезло несколько больше, чем другим. Например, родившийся в 1905 году житель американского города Сан-Бернардино Майкл Соммерс, рано созревший и постаревший, смог дожить до 31 года. Поначалу сверхбыстрое вступление во взрослую жизнь его даже радовало. Но когда в семнадцать Майкл с ужасом понял, что начал стареть, он стал предпринимать отчаянные попытки остановить этот губительный процесс.
Но врачи только разводили руками, не в силах чем-либо помочь. Немного замедлить дряхление Соммерсу удалось после того, как он, перебравшись на постоянное жительство в деревню, стал проводить много времени на свежем воздухе. Но все же к 30 годам он превратился в старика, а через год его доконал обыкновенный грипп. Среди прочих подобных феноменов можно выделить англичанку Барбару Дэлин, которая умерла в 1982 году в возрасте 26 лет.







К 20 годам успевшая побывать замужем и родить двоих детей, Барбара быстро и необратимо состарилась. Именно поэтому ее бросил молодой муж, не пожелавший жить со «старой развалиной». В 22 года от ухудшения здоровья и перенесенных потрясений «старушка» ослепла и до самой смерти передвигалась на ощупь или в сопровождении собаки-поводыря, подаренной ей властями ее родного Бирмингема.
Полю Демонжо из французского города Марселя двадцать три года. При этом выглядит он на все 60 и ощущает себя человеком преклонного возраста. Однако пока не теряет надежды, что свершится чудо и будет найдено средство, которое прекратит его стремительное одряхление. Его собрату по несчастью, сицилийцу из города Сиракузы Марио Термини нет и 20 лет лет, однако на вид ему намного больше 30. Сын богатых родителей, Термини ни в чем себе не отказывает, встречается с местными красотками и ведет разгульный образ жизни.
Во всем виноваты гены
Многие ученые считают, что основная причина этого заболевания – генетическая мутация, ведущая к накоплению большого количества протеина в клетках. Экстрасенсы и маги утверждают, что есть специальные методики насылания «порчи» с целью состарить человека.

Кстати, эта болезнь встречается не только у людей, но и у животных. У них также жизненные циклы и периоды порой идут по сценарию год за три, а то и за десять лет. Возможно, решение проблемы будет найдено именно после многолетних экспериментов на братьях наших меньших.
Как удалось установить исследователям из Калифорнийского Университета, препарат под названием «ингибитор фарнезилтрансферазы» существенно снижает скорость проявления симптомов преждевременного старения у лабораторных мышей. Возможно, это лекарство окажется пригодным и для лечения людей.
Вот как характеризует симптомы недуга у детей кандидат биологических наук Игорь Быков: «Прогерия возникает внезапно с появления крупных пигментных пятен на теле. Затем людей начинают одолевать самые настоящие старческие хвори. У них развиваются болезни сердца, сосудов, диабет, выпадают волосы и зубы, исчезает подкожный жир. Кости делаются ломкими, кожа морщинистой, а тела – сгорбленными. Процесс старения у таких больных протекает примерно в десять раз быстрее, чем у здорового человека. Зло коренится, скорее всего, в генах. Есть гипотеза, что они вдруг перестают отдавать клеткам команду делиться. И те быстро приходят в негодность».
Гены перестают отдавать клеткам команду делится вроде бы от того, что укорачиваются кончики ДНК в хромосомах, – так называемые теломеры, длиной которых предположительно и отмерен срок человеческой жизни. Подобные процессы идут и у нормальных людей, но гораздо медленнее. Но совершенно непонятно, в результате какого именно нарушения укорачиваются теломеры и начинается ускорение старения как минимум в 10 раз. Сейчас ученые с помощью ферментов пытаются удлинить теломеры. Появились даже сообщения, что американским генетикам удалось таким образом продлить жизнь мухам. Но до результатов, применимых на практике, пока далеко. Людям не удается помочь даже на уровне экспериментов. К счастью, по наследству недуг не передается.
Предполагается, что сбой в геноме происходит еще в период внутриутробного развития. Пока наука не может отслеживать и управлять этим сбоем: она может только констатировать факт, но, возможно в недалеком будущем геронтология ответит миру на этот вопрос.