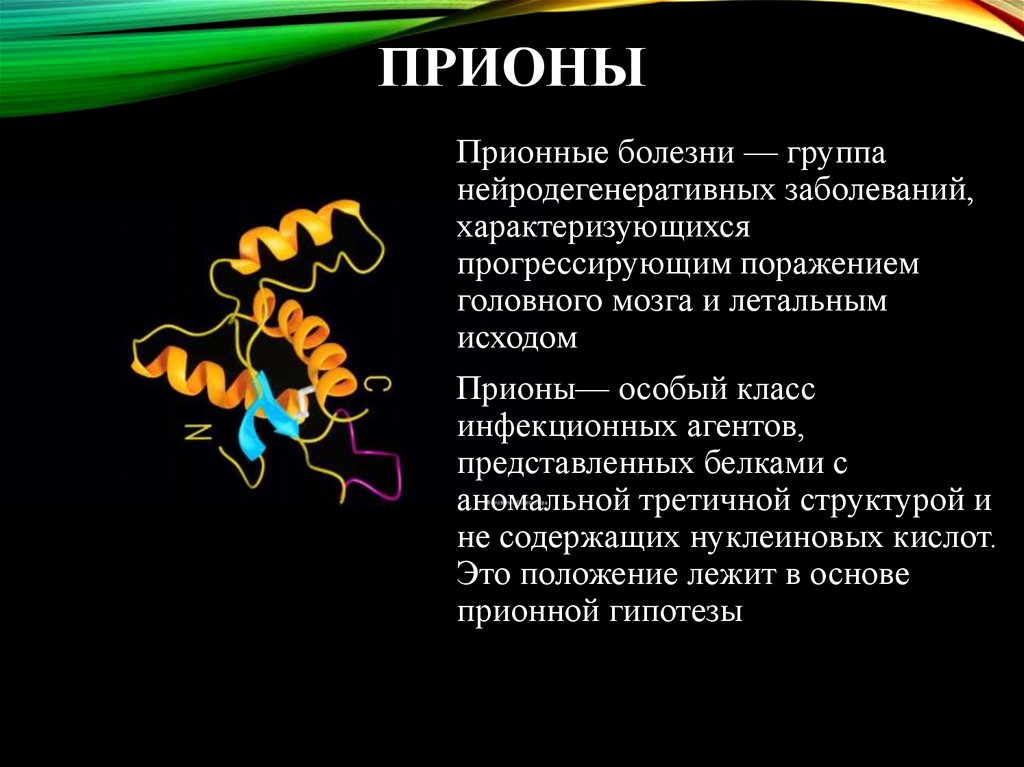
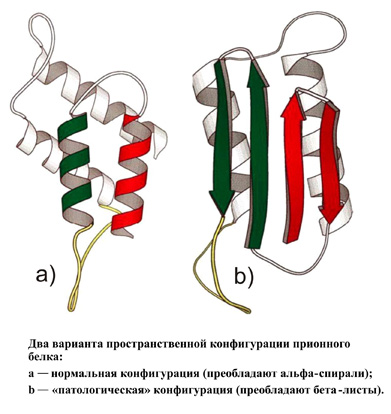
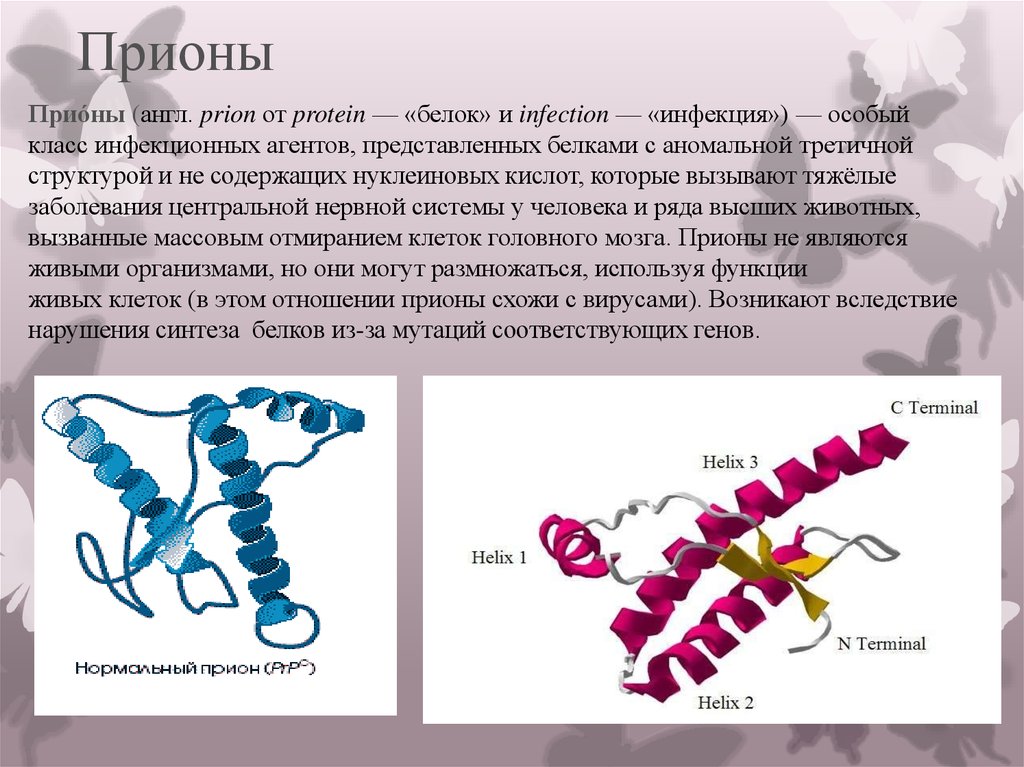
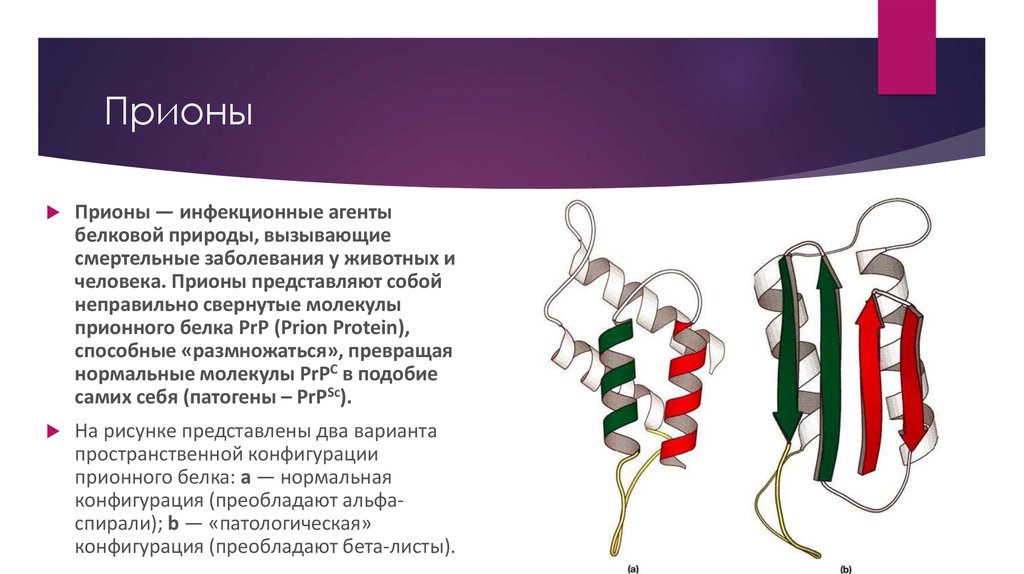
Этиология[править]
Человек может заразиться прионами, содержащимися в пище, так как они не разрушаются ферментами пищеварительного тракта. Беспрепятственно проникая через стенку тонкого кишечника, они в конечном итоге попадают в центральную нервную систему. Так переносится новый вариант болезни Крейтцфельдта-Якоба (nvCJD), которой люди заражаются после употребления в пищу говядины, содержащей нервную ткань из голов скота, больных бычьей губчатой энцефалопатией (BSE, коровье бешенство).
Прионы могут проникать в тело и парентеральным путем. Были описаны случаи заражения при внутримышечном введении препаратов, изготовленных из человеческих гипофизов (главным образом гормоны роста для лечения карликовости), а также заражение мозга инструментами при нейрохирургических операциях, поскольку прионы устойчивы к применяемым в настоящее время термическим и химическим методам стерилизации. Эта форма болезни Крейтцфельдта-Якоба обозначается как ятрогенная (1CJD).
При определенных, неизвестных условиях, в организме человека может произойти спонтанная трансформация прионового протеина в прион. Так возникает так называемая спорадическая болезнь Крейтцфельдта-Якоба (sCJD), впервые описанная в 1920 г. независимо друг от друга Гансом Герхардом Крейтцфельдтом и Альфонсом Марией Якобом. Предполагается, что спонтанное возникновение этой болезни связано с фактом, что в норме в человеческом теле постоянно возникает небольшое количество прионов, которые эффективно ликвидируются клеточным Аппаратом Гольджи. Нарушение этой способности «самоочищения» клеток может привести к повышению уровня прионов выше допустимой границы нормы и к их дальнейшему неконтролируемому распространению. Причиной возникновения спорадической болезни Крейтцфельдта-Якоба согласно этой теории является нарушение функции Аппарата Гольджи в клетках.
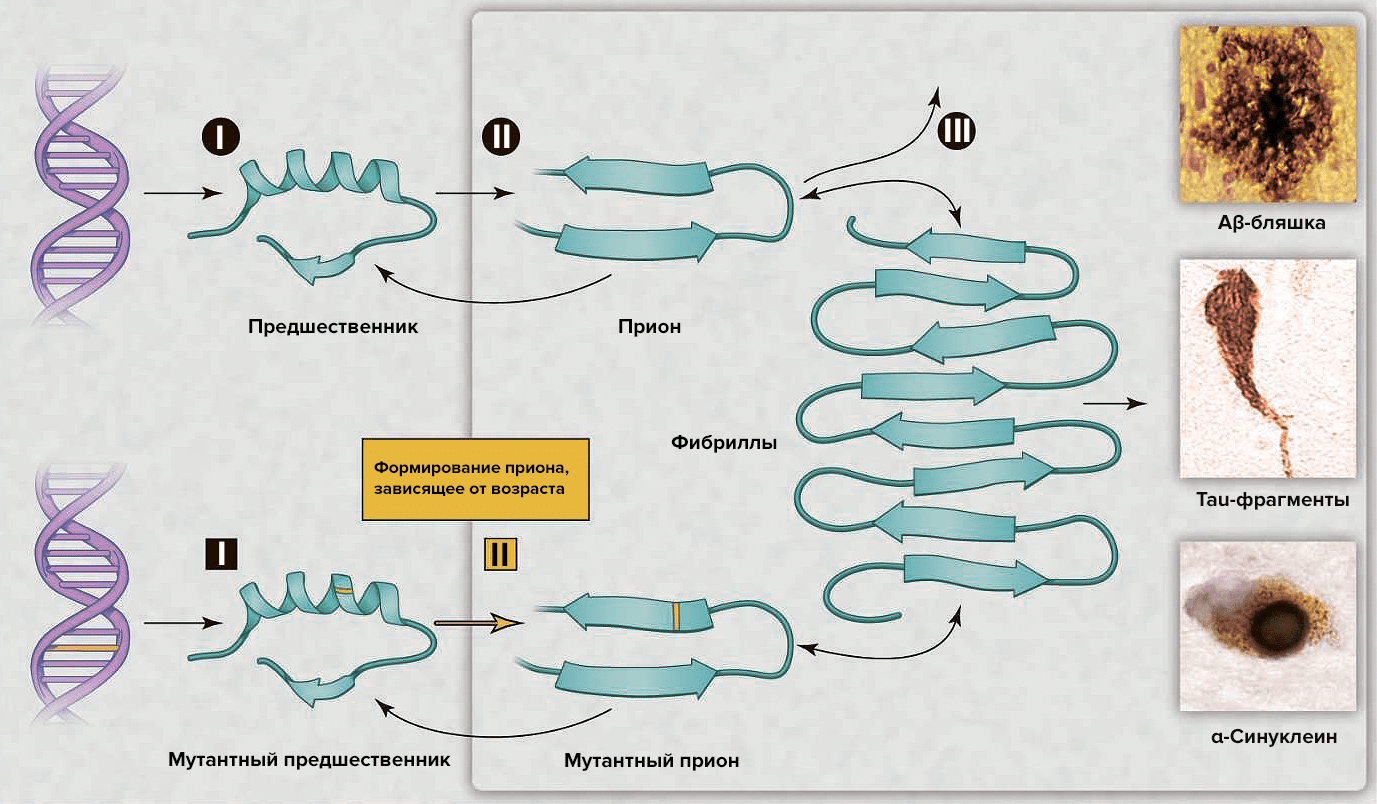
Особую группу прионовых заболеваний представляют собой наследственные (врожденные) болезни, вызванные мутацией гена прионового протеина, который делает возникший прионовый протеин более подверженным спонтанному изменению пространственной конфигурации и превращения их в прионы. К этой группе наследственных заболеваний относится и наследственная форма болезни Крейтцфельдта-Якоба (fCJD), которая наблюдается в ряде стран мира.
При прионовой патологии наивысшая концентрация прионов обнаружена в нервной ткани зараженных людей. Значительное количество прионов встречается в лимфатической ткани. Наличие прионов в биологических жидкостях, включая слюну, пока не было однозначно подтверждено. Если представление о постоянном возникновении небольшого количества прионов верно, то можно предположить, что новые, более чувствительные методы диагностики откроют это количество прионов, разбросанное по различным тканям. В данном случае, однако, речь пойдет о «физиологическом» уровне прионов, которые не представляют собой никакой угрозы для человека.
Прионы и медицинские инструменты[править]
Прионы очень стойки к обычным методам дезинфекции. Ионизирующее, ультрафиолетовое или микроволновое излучение на них практически не действует. Дезинфекционные средства, обычно используемые в медицинской практике, действуют на них лишь в очень ограниченной мере. Надежно их ликвидируют дезинфицирующие реактивы — сильные окислители, разрушающе действующие на протеины.
Другое затруднение представляет собой стойкость прионов к высоким температурам. Даже при автоклавировании при 134 °C в течение 18 минут невозможно достичь полного разрушения прионов, и прионы «выживают» в форме, способной вызвать заражение. Стойкость к высоким температурам еще более возрастает, если прионы засохнут на поверхности металла или стекла или если образцы перед автоклавированием были подвергнуты действию формальдегида.
В Великобритании, где новый вариант является очень серьезной проблемой, по этим причинам уже используются одноразовые хирургические инструменты для тонзиллэктомии. В будущем напрашивается альтернативное решение: создания новых инструментов, с учётом повышенных требований к очистке и обеззараживанию. Одноразовое использование инструментов согласно принципам ВОЗ требуется в случае стоматологического обслуживания пациентов с диагностированным прионным заболеванием или в случае подозрения на него.
Намного более сложным решением этой проблемы является лечение пациентов группы риска. К ним относятся пациенты, которые подверглись операциям, при которых была использована потенциально зараженная твердая мозговая оболочка, или пациенты из семей с наследственной формой болезни Крейтцфельдта-Якоба. ВОЗ в этом случае не требует никаких специальных мер. Британский Консультационный научный комитет по губчатой энцефалопатии в своем решении в г. счел возможным ограничиться более тщательной очисткой и обеззараживанием инструментов, в сочетании с более длительным автоклавированием.
Ты помнишь, как все начиналось
В двадцатые годы прошлого столетия врачи столкнулись с новым и неизведанным доселе заболеванием. Немецкий невропатолог Ганс Герхард Крейтцфельдт наблюдал в своей клинике одну пациентку – 20-летнюю девушку. На начальной стадии болезни у нее была нарушена чувствительность в руках и ногах, быстро прогрессировали расстройства памяти, нервной деятельности, больная все чаще впадала в бессознательное состояние. Через несколько месяцев девушка умерла от расстройств дыхания и сердечной деятельности. Невропатолог, который в будущем станет видным нацистским врачом и будет принимать участие в программе «Эвтаназия», задокументировал ход болезни.
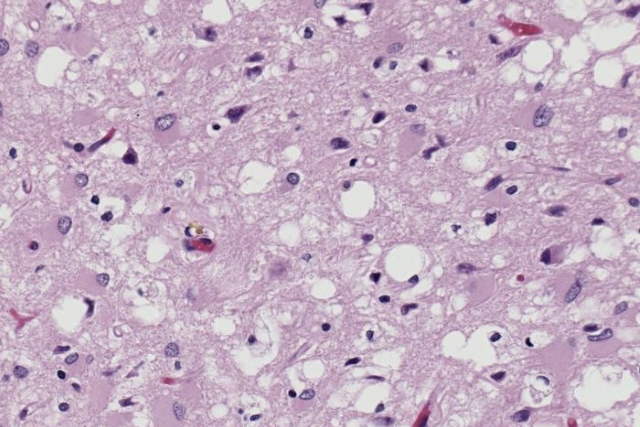
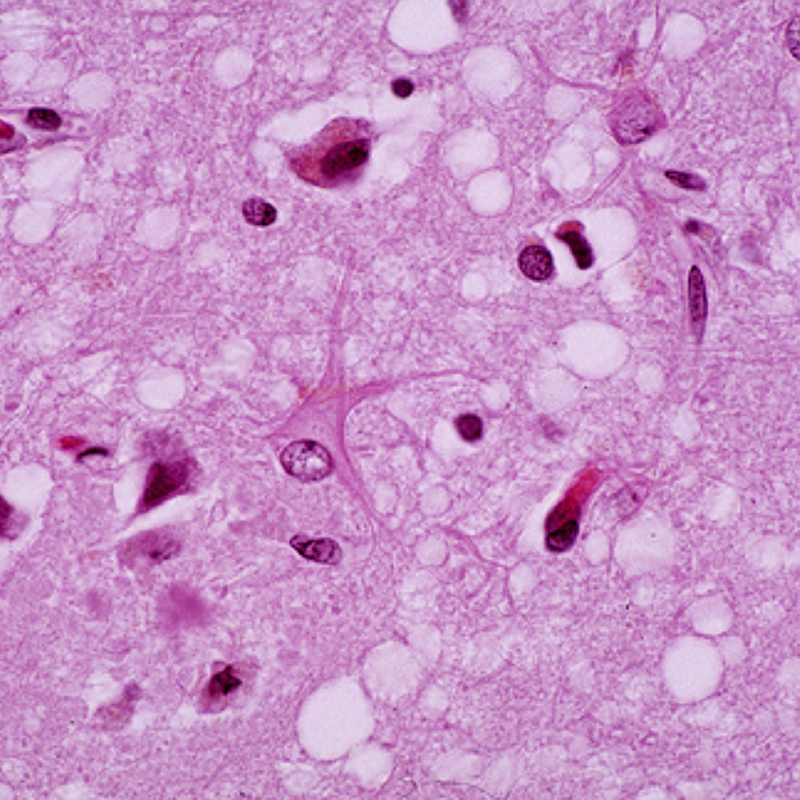
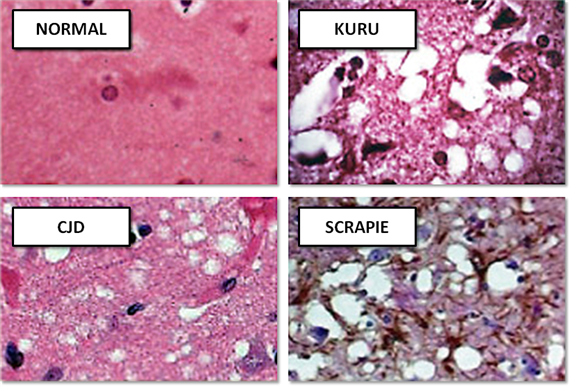
Спустя несколько месяцев доктор Альфонс Мария Якоб из Гамбурга столкнулся с тремя аналогичными пациентами. Молодые люди страдали от расстройств нервной деятельности, глотания, практически не осознавали происходящее вокруг и вскоре умерли. При вскрытии Якоб увидел интересное явление, которое раньше врачам наблюдать не приходилось, – поражен у больных был только мозг. Была зафиксирована массовая гибель клеток серого вещества головного мозга, а сохранившиеся нейроны отличались необычным набуханием. Ни в одном другом органе не было зафиксировано никаких патологических изменений. В память о двух первооткрывателях заболевание получило название болезни Крейтцфельдта – Якоба.
В те далекие годы вирусология как наука находилась еще в зачаточной стадии. Поэтому заболеванию было суждено долгое время оставаться в забвении. Этому поспособствовали Великая депрессия и Вторая мировая война. И лишь в пятидесятые годы прошлого века ученые начали активно интересоваться, что же все-таки происходит с людьми, которым не посчастливилось подхватить болезнь Крейтцфельдта – Якоба.
В то же время ученые открывают еще два заболевания, которые по своим симптомам и течению весьма и весьма напоминают описанный выше страшный недуг – куру и скрейпи. Первая болезнь была распространена среди народности форе на острове Папуа – Новая Гвинея, а вторым страдали овцы по всему миру. Но важным оказалось другое: симптомы болезней несколько отличались от болезни Крейтцфельдта – Якоба, но характер поражений был практически идентичен – образование пустот в тканях головного мозга и массовая гибель нервных клеток.


Казалось бы, все ясно. Имеется болезнь, ее вызывает какой-то вирус или бактерия, давайте разберемся, кто является возбудителем и устраним причину. Но не тут-то было! Все оказалось не так просто…
«Познавательные фильмы»: Вакцины
Прионные варианты
Прион [PSI+], как и прионы млекопитающих, может существовать во множестве вариантов. Обычно их различают по эффективности супрессии нонсенс-мутаций и стабильности наследования. Эти параметры коррелируют и определяются с невысокой точностью, что не позволяет различить с их помощью большое количество вариантов. Поэтому общее число возможных вариантов неясно, их могут быть как единицы, так и сотни.
На практике варианты [PSI+] грубо делят на два класса — сильные и слабые. Для первых характерна высокая эффективность супрессии нонсенс мутаций и высокая стабильность наследования, у вторых эти параметры снижены. Из результатов биохимического анализа следует, что клетки с сильным [PSI+] содержат меньше мономерного Sup35 по сравнению со слабым [PSI+], а прионные полимеры Sup35 более коротки. Это позволяет понять сравнительную динамику прионной полимеризации у вариантов [PSI+]. Меньший размер прионных частиц означает их большее число и более эффективную полимеризацию, что приводит к меньшему количеству мономерного Sup35 и более эффективной супрессии. Увеличенное число прионных частиц также обеспечивает более высокую стабильность наследования. В целом, «сила» варианта [PSI+] возрастает с увеличением частоты фрагментации прионных частиц [].
Примечательно, что некоторым структурно возможным вариантам амилоида Sup35 соответствуют параметры [PSI+], несовместимые либо с жизнью клетки, либо с наследованием приона. Так, в некоторых вариантах [PSI+] остается слишком мало мономерного Sup35, что нарушает жизнеспособность клетки []. На противоположном конце спектра вариантов [PSI+] находятся ненаследуемые амилоиды Sup35, обнаруженные нами. Они образуются при сверхпродукции Sup35 в клетках, содержащих прион [PIN+]. Этот прион, основанный на белке Rnq1, известен тем, что он облегчает возникновение [PSI+], видимо, служа несовершенной затравкой для полимеризации Sup35. При стандартном уровне Sup35 в клетках [PIN+] его полимеров очень мало, однако при сверхпродукции Sup35 (примерно в 20 раз) его доля в полимерной фракции возрастает до 80%. Эти полимеры, однако, неспособны к самостоятельному наследованию, и существуют лишь благодаря их частому возникновению на основе прионных частиц Rnq1. Такие полимеры Sup35 имеют очень большой размер, что говорит об их очень редкой фрагментации []. Таким образом, существующие in vivo амилоиды Sup35 можно ранжировать по частоте фрагментации: от часто фрагментируемых сильных [PSI+] до слишком редко фрагментируемых ненаследуемых.
Можно полагать, что редкая фрагментация может не только быть причиной ненаследуемости амилоидов в дрожжах, но и объяснять неинфекционность амилоидных болезней человека в сравнении с прионами. Инфекционность амилоидов должна возрастать с уменьшением их размера, поскольку при этом больше амилоидных частиц на единицу массы и выше их способность к перемещению. Прионные частицы PrPSc имеют сравнительно малый размер. Например, при болезни Крейцфельдта — Якоба прионные отложения (бляшки) часто появляются позже, чем первые симптомы заболевания, или не образуются вовсе, что указывает на малый размер приона. А интенсивное формирование бляшек при амилоидных болезнях свидетельствует о большом размере и малой мобильности амилоидных частиц, что уменьшает инфекционность. Грань между прионами и амилоидами тонка: известно, что некоторые амилоидные болезни обладают слабой инфекционностью, которую можно выявить у модельных животных при условиях, способствующих амилоидогенезу. Такими условиями могут быть повышенная продукция амилоидогенного белка, либо введение амилоидов не через пищевой тракт, а внутрицеребрально.
Добрые монстры
Такие свойства характерны не только для PrPС — даже у дрожжей найден свой «прионный белок» Ure2, способный переходить в нестандартную, но крайне устойчивую амилоидную форму. Возможно, это неспроста: «Прионы могут и не быть патогенными, — написал по этому поводу исследователь Рэндал Халфманн, — они могут играть роль «не-нуклеиновой», белковой наследственности у здоровых клеток и организмов». В подтверждение этой идеи Халфманн и его коллеги показали, что дрожжевой белок Ure2, приобретая нестандартную конформацию, влияет на целый ряд сигнальных путей клетки, в том числе и на активность гена FLO11. В свою очередь, синтезируемый на этом гене белок Flo11p необходим клеткам дрожжей для того, чтобы «сцепляться» друг с другом, формируя устойчивые к неблагоприятным условиям пленки. Так «прионное перерождение» белка Ure2 может способствовать адаптации и выживанию клеток.
Еще одну ложку меда к бочке прионного дегтя добавили работы Джеймса Чена и его коллег. Исследователи показали, что белок MAVS, один из необходимых для работы нашей иммунной системы, переходит в амилоидную прионную форму не во зло, а исключительно во благо. Обнаружив инфицированную определенными вирусами клетку, он «оседает» в ней, и его накопление служит сигналом к усиленному синтезу интерферонов и привлечению макрофагов для уничтожения зараженной клетки вместе со всем ее опасным содержимым.
«На этот счет существует интересная гипотеза, которая указывает на то, что такие амилоидные формы могут иметься вообще у всех белков, что это общее свойство полипептидных цепей, — продолжает Илья Баскаков. — Мало того, такие структуры наиболее стабильны. Поэтому высказано предположение о том, что именно в таких формах белки могли существовать в «пребиотическом супе», в котором некогда проходила химическая эволюция молекул и зарождалась жизнь». Действительно, некоторые эволюционисты полагают, что амилоидные конформации белков обладают всеми ключевыми способностями, необходимыми для роли «предков жизни». Они способны изменяться и размножаться, выполнять определенные функции, передавая свои особенности следующим поколениям. Если эта идея справедлива, то мы живем в очень странном мире, где все живое появилось на свет от странных и опасных белков, которые сегодня мы можем воспринимать не иначе как безжалостных зомби.
Статья «Зомби в клетке. Прионы: больные молекулы» опубликована в журнале «Популярная механика»
(№11, Ноябрь 2015).
Функции PrP
Одним из объяснений нейродегенерации, вызываемой прионами, может быть нарушение функционирования PrP. Однако нормальная функция этого белка изучена плохо. Данные in vitro указывают на множество разнообразных ролей, а эксперименты на мышах, «нокаутных» по этому гену, дали относительно немного информации, поскольку у этих животных наблюдались лишь малые отклонения от нормы. Недавние исследования, проведённые на мышах, показали, что расщепление PrP в периферических нервах активирует восстановление их миелинового слоя шванновскими клетками и что отсутствие PrP приводит к демиелинизации нервов.


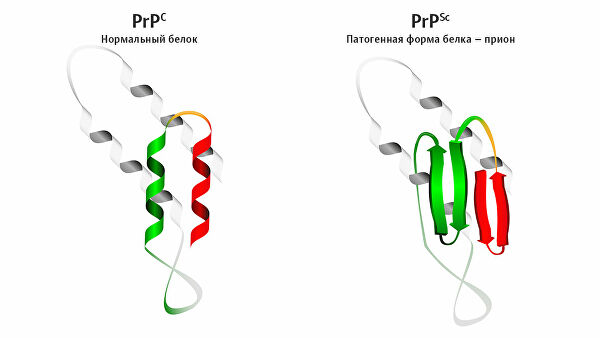
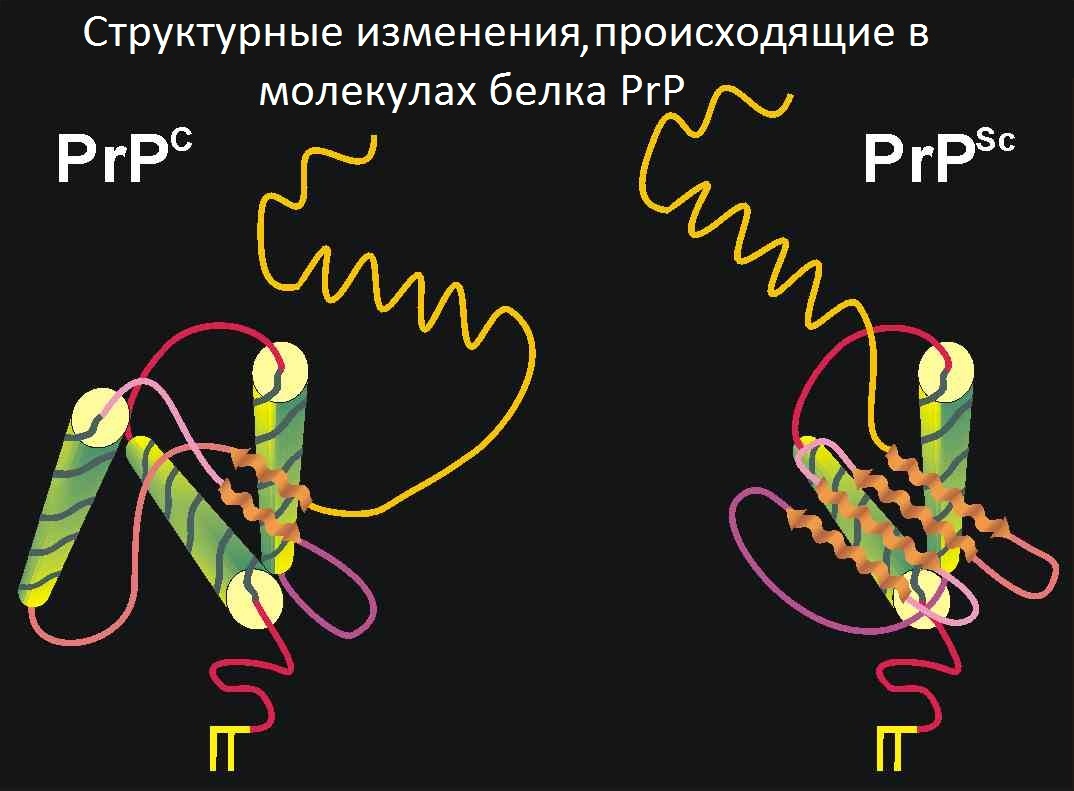
В 2005 году было выдвинуто предположение, что в норме PrP играет роль в поддержании долговременной памяти. Кроме того, у мышей, лишённых гена Prnp, наблюдается изменённая гиппокампальная долговременная потенциация.
В 2006 году учёные из Института биомедицинских исследований Уайтхед показали, что экспрессия гена Prnp в гемопоэтических стволовых клетках необходима для самоподдержания костного мозга. В исследовании было выявлено, что долгоживущие гемопоэтические стволовые клетки несут PrP на клеточной мембране, а кроветворные ткани со стволовыми клетками, лишёнными PrP, имеют большую чувствительность к клеточному истощению.
Можно ли защитить здоровье человека?
Организм человека сам производит белок PrP, неправильная конфигурация которого приводит к смертельному заболеванию. Почему бы не избавиться от этого белка? Если найти способ «выключить» ген, который его кодирует — человечество и иные живые существа, подверженные прионным заболеваниям, были бы надежно защищены от них. Но нет. Сделать это невозможно по той простой причине, что данный ген и данный белок жизненно необходимы. При их отсутствии (или «отключении») вокруг нервных волокон перестает формироваться миелиновый слой — своего рода, изолирующая защитная оболочка для отростков нервных клеток. А это приводит к болезням периферической нервной системы и, как следствие, к параличам.
Ссылки[править]
На английском языкеправить
- Mad Cow Disease Инфорация о коровьем бешенстве, Center for Global Food Issues.
- Madcowering A BSE-TSE blog.
- The Pathological Protein — Mad Cow, Chronic Wasting, and Other Deadly Prion Diseases (2003, updated online 2005). Philip Yam, Scientific American magazine writer and News Editor.
- Прионные заболевания (2003). Dr. Sean Heaphy, Leicester University.
- Prion Diseases and the BSE Crisis (1997). Статья Stanley Prusiner — первооткрывателя прионов, из Science magazine.
- Britannica Nobel: Прион, 1997
- ICTVdb 90.001.0.01. Mammalian Prions
- Официальная страница сайта о болезни коровьего бешенства Mad Cow Disease
- News & Views on Mad Cow Disease, Mad Deer Disease, Chronic Wasting Disease, and Bovine Spongiform Encephalopathy
- Biography of Dr Prusiner
- Science Daily статья о вакцине против прионных заболеваний
- Science Daily article on transmission of prions through soil
- Хороший обзор по прионам из Science Creative Quarterly
| Эта статья должна быть полностью переписана.На странице обсуждения могут быть пояснения. |
eo:Priono
hu:Prion
ia:Prion
Как можно заразиться прионами?

На сегодняшний день известны три способа заражения:
- трансмиссивный: при передаче приона от одного вида млекопитающего к другому. Причем если ранее говорилось о существовании межвидовых барьеров, то есть передача от коровы или овцы к человеку отрицалась, то на сегодняшний день считается, что прионы могут передаваться от любого инфицированного животного или человека (теоретически), и доказана возможность заражения человека от коров и другого человека. Причиной попадания патологического приона может быть употребление в пищу мяса больного животного, использование биологических тканей животных и человека (препараты крови, пересадка роговицы или твердой мозговой оболочки и тому подобное). Считается, что различные биоматериалы обладают разной степенью патогенности. Так, наиболее заразными являются ткани мозга и мозговых оболочек ввиду наибольшего содержания в них прионов, затем – кровь и ее препараты. Попадание возбудителя под кожу и через рот (с пищей) относят к наименее заразным способам. Это означает определенную дозозависимость прионных заболеваний;
- наследственный: когда заболевание развивается в результате генной мутации, возникающей в определенной области 20-й хромосомы. Особый участок 20-й хромосомы человека отвечает за наличие в организме нормального прионного белка. Функции его до конца так и не изучены. При возникновении генных нарушений вместо нормального приона синтезируется патологический, вызывающий заболевание;
- спорадический: спонтанное возникновение аномального белка в организме человека.
Таким образом, становится понятно, что прионные заболевания могут быть как инфекционными, так и наследственными. Каким бы путем прион не возник в организме человека, он может стать причиной заражения другого человека. Это означает, что даже случайным образом возникшая прионная болезнь может передаться другому человеку трансмиссивно. Предполагается различная степень инфекционности прионов, возникших в результате мутации или спонтанно, а также полученных от другого вида млекопитающих.
Этиология (причины) прионной инфекции
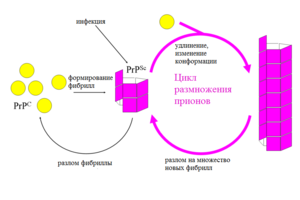
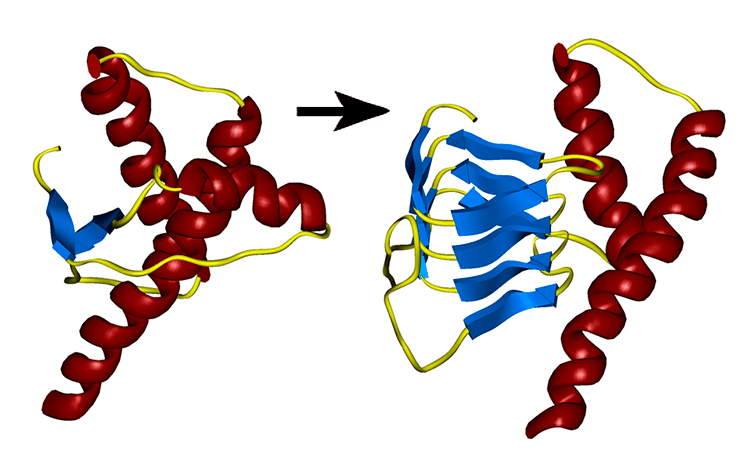
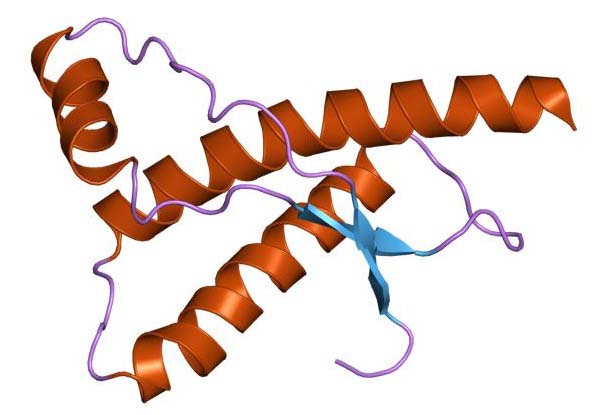
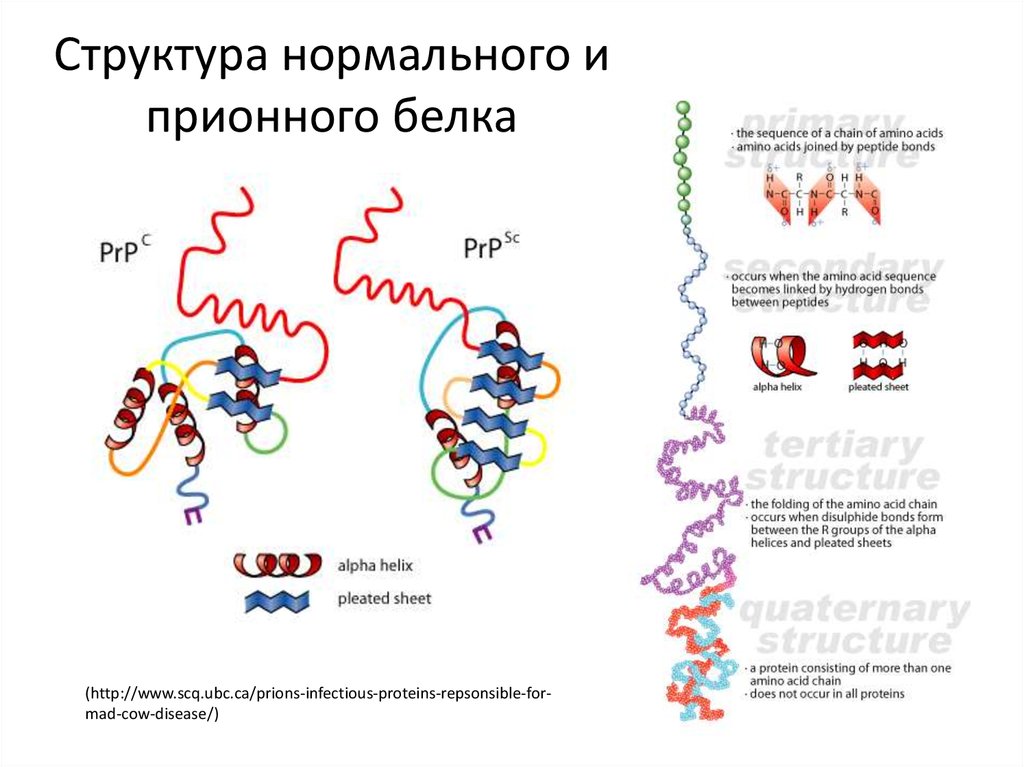
Организм человека состоит в большей степени из белков. Они являются синтетическим материалом для построения клеточных мембран, внутриклеточных структур, большинства ферментов и гормонов. Информация о том, какой белок будет синтезироваться в клетке, содержится в геноме (ДНК или РНК) и определяется последовательностью нуклеотидов. Свойства белка в свою очередь зависят от последовательности его аминокислот. После синтеза  белковой молекулы со строго заданным количеством и последовательностью аминокислот, происходит упаковка линейной молекулы белка в трехмерную структуру. Прионы – это белки с измененной трехмерной структурой. Нормальный их аналог представляет собой белок, входящий в состав нейроцитов (клетки нервной системы). Именно поэтому прионная инфекция поражает нервную систему – измененные белки, нарушают нормальную структуру клеточных мембран и приводят к образованию скоплений аномальных белков в межклеточном веществе с разрушением (дегенерация) нейроцитов. Белковые молекулы прионы обладают рядом свойств, которые приводят к развитию патологии и дают ей сходство с любой другой инфекцией:
белковой молекулы со строго заданным количеством и последовательностью аминокислот, происходит упаковка линейной молекулы белка в трехмерную структуру. Прионы – это белки с измененной трехмерной структурой. Нормальный их аналог представляет собой белок, входящий в состав нейроцитов (клетки нервной системы). Именно поэтому прионная инфекция поражает нервную систему – измененные белки, нарушают нормальную структуру клеточных мембран и приводят к образованию скоплений аномальных белков в межклеточном веществе с разрушением (дегенерация) нейроцитов. Белковые молекулы прионы обладают рядом свойств, которые приводят к развитию патологии и дают ей сходство с любой другой инфекцией:
- При попадании в клетку, они постепенно приводят к изменению упаковки синтезированного белка в правильную трехмерную структуру.
- Обладают способностью к распространению и увеличению количества аномальных белковых молекул – этот процесс происходит постоянно, в определенной прогрессии.
- Имеют возможности к заражению – процесс начинается после попадания в клетку прионов.
В отличие от инфекций, представителей живой материи, прионы являются очень устойчивыми в окружающей среде. Они не разрушаются под действием дезинфицирующих веществ, ультрафиолетового или ионизирующего облучения, высокой температуры.
Эти механизмы и особенности прионов приводят к развитию дегенеративно-дистрофической патологии нервной системы, в первую очередь головного мозга.
Прионы до сих пор вызывают оживленную дискуссию, относительно их положения в природе. Отсутствие генетического материала относит их к неживой материи. Тем не менее, способность к заражению и увеличению количества патологических белковых молекул, указывает на схожесть прионов с живой инфекцией.
Вечная молодость или неизбежная смерть
Сейчас заговорили и о том, что с прионным механизмом могут быть связаны, например, болезнь Альцгеймера или болезнь Паркинсона. Если это так, то можно ожидать появления новых методов лечения или профилактики этих серьёзных заболеваний. Интересно, что на прионы по механизму действия похожи и дрожжевые белки — вполне возможно, что дрожжи помогут в изучении этих странных молекул. Возникают и разговоры о том, что именно прионы помогут найти лекарство от разных злокачественных опухолей или ВИЧ-инфекции, да и вообще станут «философским камнем», который приведёт человечество к вечной жизни или вечной молодости. Впрочем, пока эти высказывания необоснованны.
Рустам Зиганшин поясняет, что, как и любые исследования в области биологии, изучение прионных белков расширяет границы наших знаний о живом. С практической точки зрения результаты могут помочь разобраться с тем, как развивается ряд опасных, пока ещё неизлечимых, заболеваний. Возможно, мы получим и инструмент для борьбы с ними. Если понять, как из нормального белка получается аномальный, и научиться контролировать этот процесс, то, может быть, мы сумеем запускать его и в обратную сторону — то есть превращать патологические белки в здоровые, а с ними и возвращать здоровье тканям и органам.


Фотографии: Flickr, ibreakstock — stock.adobe.com
Как уберечься от прионных инфекций
Нет способов обезопасить себя от развития болезней, вызванных прионами. Теоретически человек может пройти диагностику клеток и выяснить, есть ли у него генетические нарушения и предрасположенность к прионной болезни. Но лаборатории, способные провести такие анализы в основном расположены за рубежом, в высокоразвитых странах.
В принципе, человек может хоть как-то себя защитить от БКЯ, если исключит из употребления мясо и птицу, привезенную из «опасных» районов (мест, где были заражения скота). В основном это Европа. Кроме того, нужно отказаться от приема препаратов, в основе которых есть кровь животных или человека, лекарства нужно сменить на синтетические. Также стоит отказаться от переливания крови (если это возможно).
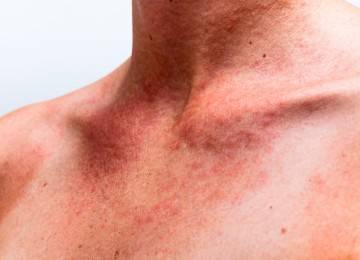
Новый вариант БКЯ: заразиться, съев кусок мяса

Впервые о возникновении этого нБКЯ предположили в 1996 году. Дальнейшие исследования подтвердили опасения ученых. Возможно, больные ели мясо, в котором содержались прионы мозга коров.
нБКНЯ не имеет конкретного возраста заражения. Инфекция вызывает у зараженных и психологические изменения (нередко это расстройство по ошибке диагностируют как психическое, вместо неврологического). Развитие болезни длится 6-8 месяцев, но иногда может достигать и полутора лет, поэтому врачам сложно поставить правильный диагноз после первых симптомов. Обычно новую форму диагностируют, когда больной перестает контролировать движения.
Последняя стадия нБКЯ проходит с быстрой потерей мышечного контроля. Пациент теряет возможность двигаться и говорить (акинетический мутизм).
Деменция и БКЯ: кто-то болен, но об этом не подозревает

Деменции с быстрым прогрессированием могут привести к смерти через несколько дней или месяцев с начала своего проявления. Примерно у 47% больных со спонтанной формой БКЯ обнаружена быстро прогрессирующая деменция, с генетической – 13%, а иные случаи относят к остальным деменциям.
Прионный белок может развиться как спонтанно, так и генетически, поэтому практически все пациенты долгое время об этом не знают.
Окончательный диагноз быстро прогрессирующей деменции ставится после дифференциальной диагностики. Но даже при тщательном обследовании больных некоторые диагнозы ставятся только после вскрытия.
Фатальная семейная бессонница

Эта болезнь не позволяет человеку спать, и лишает его возможности выполнять привычные функции. Есть спорадическая и генетическая формы. Генетическая приводит к мутации PrP белка и его преобразование в прионный. Спорадическая форма обнаруживается случайно, без наличия предпосылок. Это заболевание поражает таламус (отдел мозга, отвечающий за сон), поэтому зараженный теряет способность спать.
В среднем, первые симптомы могут появиться у пациента в 40 лет. Поначалу у больного появляются проблемы со сном и судороги. Также во время сна зараженный может непроизвольно двигать конечностями. В результате пациент вообще теряет способность спать, вследствие чего понижается активность мозга и теряется координация мышц.




Для точного диагноза генетической формы нужно провести соответствующие тестирования. Спорадическую форму можно диагностировать по нарушениям в структуре сна. В среднем, человек живет до 3 лет с момента проявления начальных симптомов.
Медленно прогрессирующая деменция — симптомы за несколько лет
Это заболевание впервые описали более 80 лет назад. В основном синдром развивается у людей среднего возраста (от 40 до 50 лет), и он вызывается мутацией на 20 хромосоме.
Симптомы заболевания примерно такие же, как со спонтанной формой БКЯ. Но деменция прогрессирует годы. Этот синдром может длиться от нескольких месяцев до нескольких лет. В среднем, пациенты живут 5 лет. Заболевание обнаруживается с помощью МРТ.
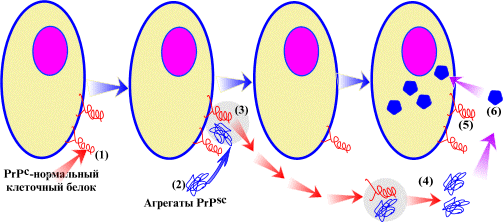
Механизм фрагментации прионов дрожжей
Ключевую роль во фрагментации прионных амилоидов в дрожжах играет Hsp104, но при существенном содействии других шаперонов. Напомним, что белки этого класса обеспечивают и контролируют правильную пространственную укладку других белков. Фрагментация начинается с того, что шаперон Sis1 семейства Hsp40 распознает в прионной частице какие-то элементы неправильно уложенной белковой структуры. Другие дрожжевые Hsp40, в частности, наиболее высоко экспрессируемый Ydj1, видимо, не участвуют во фрагментации. Согласно имеющимся данным, прионный домен упакован в амилоидную структуру не полностью, и какая-то его часть остается неструктурированной. При этом у сильных вариантов [PSI+] размер неуложенной области больше, чем у слабых. Вероятно, эту область и распознает Sis1, а затем, при участии шаперона Ssa (семейства Hsp70), передает ее Hsp104 — большому белку массой более 100 кДа, содержащему два АТФ-расщепляющих блока. Его рабочая форма представляет кольцо из шести молекул. Hsp104 цепляет полипептидную цепь прионного белка и протягивает ее сквозь дырку в кольце, таким образом расплетая этот белок и извлекая его из полимера, который в результате распадается на две части (рис. 2).
Частота фрагментации — ключевой фактор, определяющая число прионных частиц, эффективность прионного превращения и выраженность прионного фенотипа. Но что определяет частоту фрагментации?
Было обнаружено, что фибриллы, соответствующие сильному варианту [PSI+], значительно менее прочны, чем фибриллы слабого [PSI+]. В согласии с этим, картирование области Sup35, составляющей ядро (стебель) амилоида с помощью водородно-дейтериевого обмена показало, что у «сильных» фибрилл в ядро амилоида, уложена меньшая часть прионного домена (аминокислотные остатки 1–37, полный домен состоит из первых 123 остатков), чем у «слабых» (остатки 1–70) []. Эти наблюдения породили предположение, что частота фрагментации определяется прочностью амилоида.
В противоположность этому, мы выяснили, что частота фрагментации определяется в первую очередь способностью шаперонов распознавать амилоид как субстрат для фрагментации. В частности, «сильную» фибриллу Sup35 легче узнать, чем «слабую», поскольку у нее больше неструктурированный остаток прионного домена, не уложенный в ядро и привлекающий шапероны []. Но как распознаваемость зависит от последовательности прионного домена? Помимо размера неупорядоченной области, все зависит от ее аминокислотного состава, но не требует каких-либо специфических элементов последовательности, поскольку, как было установлено, ее случайная перетасовка в прионном домене Sup35 не нарушает его прионных свойств []. Чтобы изучить влияние аминокислотного состава на фрагментацию, мы создали набор генов Sup35, в которых прионный домен был заменен на последовательности (QQQXQ)n, где Q — глутамин, а Х — любая аминокислота, с общей длиной повторов от 70 до 100 остатков. Полимеры на основе чисто полиглутаминовой последовательности фрагментируются достаточно редко, что позволяет наблюдать эффект добавления других аминокислот. Например ароматические аминокислоты тирозин, триптофан или фенилаланин вызывали максимальный эффект, многократно усиливая фрагментацию, а аланин, серин, треонин, цистеин и метионин — средний. Аспарагин, глицин, валин и изолейцин не улучшали фрагментацию, а пролин и лейцин препятствовали образованию полимеров []. Таким образом, частота фрагментации зависит от размера и аминокислотного состава неструктурированной области прионного домена, не упакованной в ядро амилоида.
Способы заражения
В заключение поговорим о способах заражения. Их четыре. В первом и самом распространенном случае заболевание возникает как бы из ниоткуда. То есть жил себе человек, да вдруг взял и заболел. Этот путь возникновения болезни называется спорадическим и, кстати сказать, является наиболее распространенным. По нынешним представлениям, это происходит спонтанно под действием каких-то пока не установленных факторов.
Второй способ – наследственный. Некоторые виды болезней являются семейными и возникают из-за мутаций. В свою очередь, гены передаются потомству. Известно около 40 семей, страдающих фатальной бессоницей. Каждый десятый страдающий болезнью Крейтцфельдта – Якоба – страдает семейной формой этого заболевания.
Фото: M24.ru/Михаил Сипко
Третий способ – ятрогенный. Это означает, что заражение прионами произошло по вине медицинских работников при проведении каких-либо оперативных вмешательств. Однако описаны лишь несколько таких случаев, и все они произошло в 70-е годы прошлого века, когда о свойствах прионов еще никто не знал. Так, одна женщина заболела после того, как ей пересадили роговицу глаза от страдавшего болезнью Крейтцфельдта – Якоба мужчины.
А вот последний способ наиболее коварен и опасен. Дело в том, что человек восприимчив к прионам, которые поражают крупный рогатый скот. И при употреблении в пищу мяса больных животных заболевают и люди – у них развивается болезнь Крейтцфельдта – Якоба. В девяностые годы прошлого века настоящая эпидемия этого страдания разразилась в Англии.
Лечения пока нет. Однако ученые уже выяснили, что некоторые виды прионов разлагаются лишайниками, другим удалось описать особые антиприонные антитела (к инфекционным прионам).




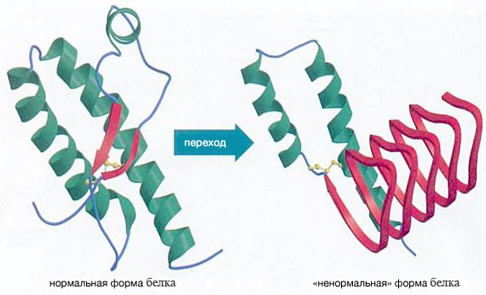


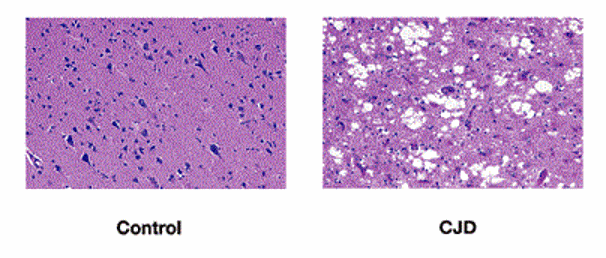
Иными словами, перед исследователями стоит весьма непростая задача, которая не только поможет найти лекарство от тяжелых заболеваний, но и, возможно, поможет открыть секрет долголетия. Для этого нужно только одно – понять прионы.
Сюжет:
От морей к звездам: как работает наука
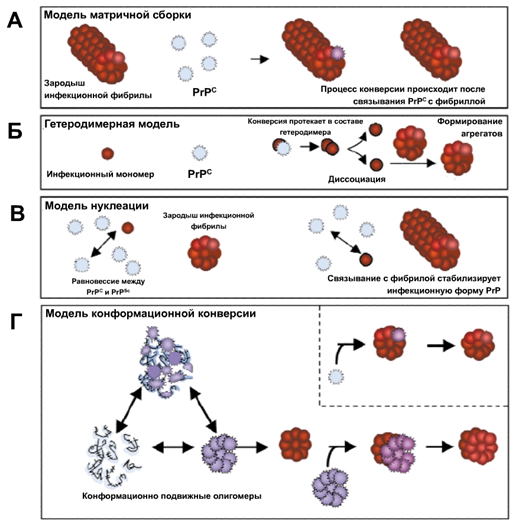
Мономер или полимер?
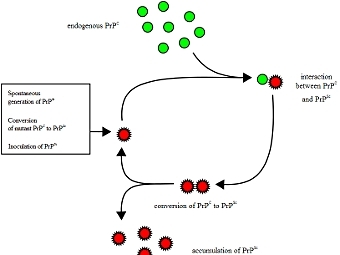
Известны две модели прионного превращения, которые полагаются на различные представления о природе этой формы белков. Первая модель [] пришла вместе с прионной концепцией для PrP, которая помогла связать загадочные детерминанты дрожжей с прионным явлением. В соответствии с этой моделью, известной как «гетеродимерная», прионная форма белка представляет собой мономер с измененной структурой, а превращение белка происходит в результате взаимодействия прионной и неприонной молекул, после чего две прионные молекулы отделяются друг от друга.
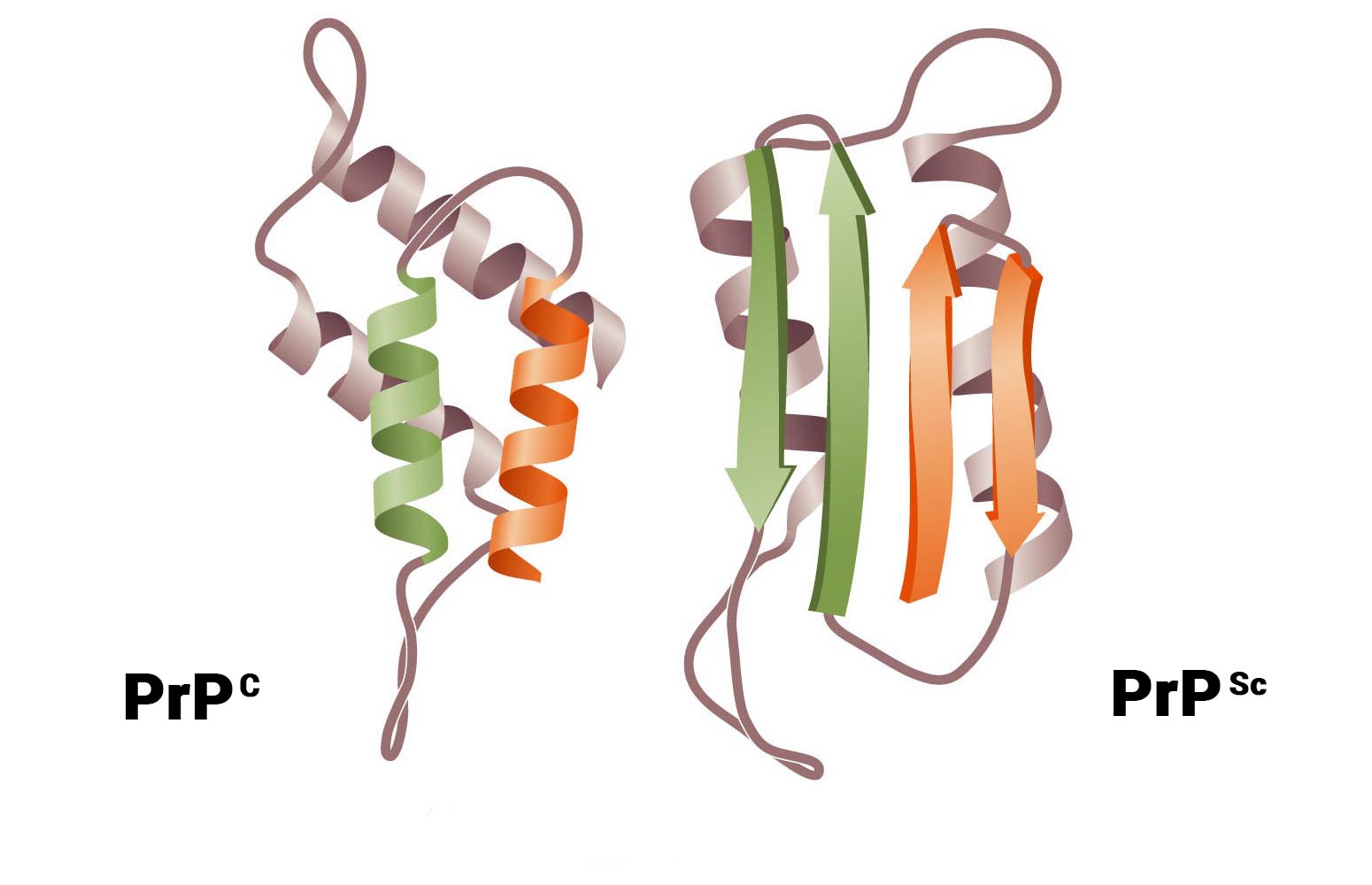
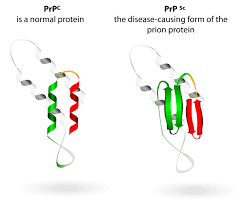
Альтернативная модель предполагает, что прион — это разновидность амилоида, т. е. нековалентно-связанный белковый полимер. Амилоидами называют фибриллярные белковые агрегаты с регулярной структурой, признаваемые причиной более 30 неизлечимых возрастных заболеваний, таких, например, как болезни Альцгеймера и Паркинсона []. Амилоиды подобны прионам по двум ключевым свойствам. Во-первых, укладка белка в составе амилоида существенно изменена и образует характерную структуру, называемую кросс-бета, в которой полипептидные цепи перпендикулярны оси фибриллы, а образуемые ими бета-слои параллельны оси. Во-вторых, амилоид катализирует структурное превращение и полимеризацию нормальной мономерной формы соответствующего белка.
Нам стало ясно, что верна именно вторая модель. Гетеродимерная модель имеет существенные слабости. Она предполагает довольно неправдоподобные свойства белка PrP. Наличие у белка двух альтернативных укладок само по себе необычно, но еще труднее представить, что одна из этих укладок способна превращать другую в себе подобную. Кроме того, известно, что существуют штаммы приона с различными свойствами, а, значит, альтернативных самовоспроизводящихся укладок должно быть множество — и это уже совершенно непредставимо. Не менее трудно объяснить и разделение двух прионных молекул после превращения одной из них, при условии, что процесс не расходует энергию в виде АТФ или ГТФ. В полимерной же модели таких проблем нет: наличие различных самовоспроизводящихся укладок в составе полимера хорошо известно, а разделение молекул в ходе полимеризации не требуется.
Следует заметить, что под формальное определение приона попадает как минимум одно явление, не связанное с амилоидами. Так, дрожжевой прион представляет собой вакуолярную протеазу В, которая синтезируется неактивной и активируется за счет удаления аминоконцевого пептида другой, активной, молекулой протеазы В. Затем наша молекула может активировать другие молекулы протеазы В, удаляя пептид, что, кстати, напоминает гетеродимерную модель [].