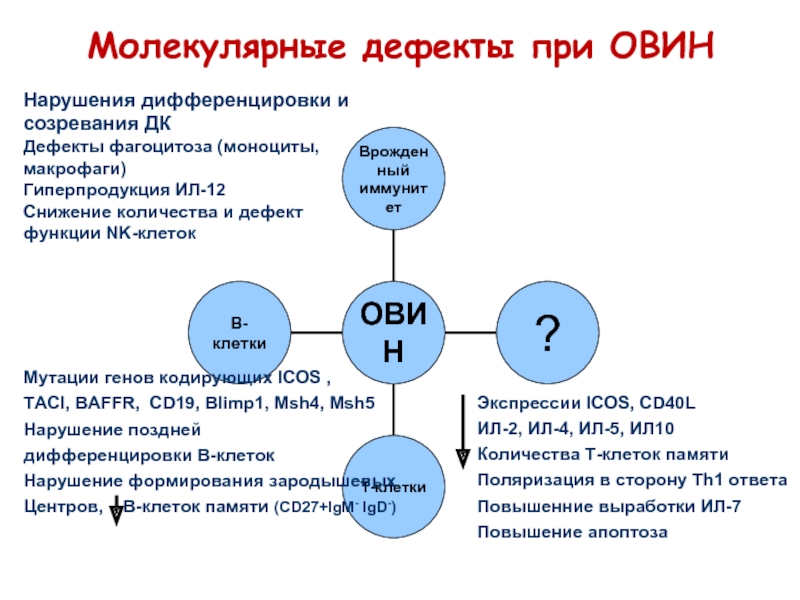
Клинический случай
Мальчик С., 4,5 года, госпитализирован в отделение интенсивной терапии по поводу 4-го эпизода тяжелой пневмонии, осложненной емпіємою плевры.
Ребенок родился доношенным от первых физиологических родов, течение беременности – неосложненный. Мальчик развивался нормально до 8 месяцев жизни на грудном вскармливании находился до 6 месяцев. Получил вакцинацию БЦЖ, АКДС и живую полиомиелитную вакцину, осложнений не было. Впервые госпитализирован в возрасте 8 месяцев по поводу абсцедирующие фурункула левого бедра, лечение – оперативное с курсом антибактериальной терапии. Рецидивирующий фурункулез наблюдался на втором году жизни, мальчик получил несколько курсов лечения антибиотиками.В возрасте 2 лет госпитализирован впервые с проявлениями левосторонней нижньодолевої пневмонии. Заболевание характеризовалось затяжным течением, проведено бронхоскопию, выявлен гнойный эндобронхит. Через полгода – повторный эпизод пневмонии с тяжелым затяжным течением. На 4-м году жизни – две госпитализации в отделение интенсивной терапии по поводу пневмонии, осложненной гнойным плевритом.
При обследовании выявлено гипоплазию миндалин, гипоплазию лимфатических узлов. Показатели роста и веса были несколько снижены по сравнению с возрастной нормой.
В результате лабораторного обследования при госпитализации диагностирована тяжелую нейтропению: лейкоциты 4,0х109/л, пал. 1%, сегм. 4%, лимф. 85%, эоз. 5%, мон. 10%, абсолютное количество нейтрофилов – 200/мкл (норма – более 1000/мкл). Динамика показателей гемограммы была положительной на фоне проведенного лечения. Массивную антибактериальную терапию проводили в течение месяца, клиническое состояние мальчика и показатели гемограммы постепенно нормализовались. При иммунологическом обследовании выявлен чрезвычайно низкий уровень сывороточных иммуноглобулинов: ІgG – 0,45 г/л (возрастная норма – 9,29 ± 2,28 г/л), Ида – 0 (0,93 + 0,27), ІдМ – 0 (0,56 + 0,18).
Учитывая повторные инвазивные бактериальные инфекции, гипоплазию лимфоидной ткани, чрезвычайно низкие уровни сывороточных иммуноглобулинов у пациента был заподозрен первичный иммунодефицит, предположительно Х-сцепленный. При исследовании субпопуляций лимфоцитов методом проточной цитофлюорометрії с применением моноклональных антител выявлено почти полное отсутствие В-лимфоцитов.
Дефицит В-лимфоцитов характерен для Х-сцепленной формы агаммаглобулинемия, при которой отсутствие Вtk (Bruton’s tyrosine kinase) нарушает нормальное развитие В-клеток. Нормальное количество Т-лимфоцитов у пациента позволяет исключить Х-сцепленную форму тяжелого комбинированного иммунодефицита, при которой отсутствуют Т-клетки, В-клетки представлены в нормальном количестве. При гипер-ІдМ синдроме количество В-лимфоцитов также соответствует норме, характерен высокий или нормальный уровень ІдМ при отсутствии ІgG и Ида.
Следует отметить, что нейтропения нередко ассоциируется с различными формами гипогаммаглобулинемии, поэтому у детей с нейтропенией и рецидивирующими инфекциями показано определение уровня иммуноглобулинов (табл. 2, 3) . Нейтропения может рецидивировать, в частности часто сочетаться с эпизодами тяжелых бактериальных инфекций, что и было выявлено у пациента.
Через некоторое время после выписки из стационара у мальчика появились симптомы артрита левого коленного сустава, что также является достаточно характерным проявлением Х-сцепленной агаммаглобулинемия и описано у 30-50% больных первичным иммунодефицитом .
Осложнения
Протекание инфекционных болезней характеризуется тяжелым состоянием больного. Зачастую инфекции сопровождаются различными осложнениями.
Для агаммаглобулинемии характерны такие осложнения как:
-
постоянно повторяющиеся тяжелые инфекционные заболевания;
-
возникновение хронических очагов болезней;
-
начало заболеваний, направляющих работу иммунной системы против организма;
-
появление различных болезней ЖКТ, нервной системы;
-
повышенный риск возникновения раковых опухолей.
В запущенной стадии и без необходимого лечения болезнь Брутона приводит к летальному исходу из-за многочисленных инфекционных заболеваний.
Лечение агаммаглобулинемии
Терапия агаммаглобулинемии – консервативная, некоторые назначения выполняются до конца жизни. Лечение этой патологии бывает:
- общее;
- местное.
Общими назначениями являются:
- правильная диета для укрепления общих иммунных сил организма. Это должно быть полноценное в широком понимании питание – с достаточным количеством белков, жиров, углеводов. Такие пациенты должны избегать диет для похудения, а если в этом возникла потребность – то соблюдать их только после консультации и под наблюдением диетолога и врача, который занимается проблемой агаммаглобулинемии у конкретного пациента;
- иммуноглобулин либо донорская плазма от здоровых доноров. Сперва проводится курс лечения, далее в качестве поддерживающей терапии пациенту на протяжении всей жизни регулярно вводят определенное количество препарата;
- антибактериальные средства. До получения микробиологических результатов назначают антибиотики широкого спектра действия, далее – в зависимости от выявленной чувствительности конкретного возбудителя. Как правило, назначают антибиотики пенициллинового ряда, макролиды, цефалоспорины, аминогликозиды;
- нестероидные противовоспалительные препараты – их назначают по потребности в зависимости от инфекционно-воспалительного поражения на фоне агаммаглобулинемии. Так, НПВП показаны при поражении ЛОР-органов, но из-за раздражающего действия на слизистую оболочку пищеварительного тракта их применения следует избегать при болезнях ЖКТ.
Местное лечение проводится в зависимости от пораженного органа – как правило, при ЛОР-патологиях. Выполняются:
- санирование дополнительных пазух носа антисептическими растворами;
- полоскание горла
и так далее.
Развитие патологии
Следует различать врожденные и генетические болезни (в данном случае – иммунной системы). Первое означает, что патология у человека проявляется уже с рождения, он ее «приобрел» еще во время внутриутробного развития из-за воздействия каких-то патогенных факторов. Второе означает, что нарушения появляются из-за сбоя генов, кодирующих те или иные признаки и свойства органов и тканей, но такие сбои могут наступать не только при передаче родительского генного материала ребенку, а и вследствие мутаций после рождения. Агаммаглобулинемия относится к числу тех патологий, при которых из-за мутаций, затронувших переданные гены, нарушение формируется еще внутриутробно.









Данное заболевание может передаваться двумя путями – поврежденными половыми и неполовыми хромосомами.
Существуют три разновидности наследственной агаммаглобулинемии:
- сцепленная с X-хромосомой;
- аутосомно-рецессивная спорадическая швейцарского типа;
- сцепленная с X-хромосомой и связанная с нехваткой гормона роста.
Обратите внимание
Чаще всего диагностируется та форма агаммаглобулинемии, которая сцеплена с X-хромосомой – из 10 пациентов с агаммаглобулиемией данную ее разновидность диагностируют у 8-9. Но ею болеют только представители мужского пола. Также только у мальчиков встречается агаммаглобулинемия, сцепленная с X-хромосомой и связанная с недостаточностью гормона роста.
У представителей обоих полов диагностируют только аутосомно-рецессивную спорадическую агаммаглобулинемию швейцарского типа.
Что происходит при патологии, связанной с нарушением половой хромосомы – так называемой X-сцепленной форме? При этом изменена структура гена, который отвечает за правильную выработку фермента, ответственного за формирование и дифференцировку (специализацию) B-лимфоцитов, которые являются клетками иммунной системы. Этот фермент буквально блокируется. Как результат, нарушается так называемый гуморальный (клеточный) иммунитет.
Суть патологии на клеточном уровне такова. Предшественники B-клеток успешно появляются – их можно выявить в костном мозге. Но далее процесс их «специализации» (ответственности за одно из звеньев иммунитета) прекращается, равно как и поступление в кровь. Из-за того, что блокируется синтез всех классов иммуноглобулинов, в иммунитете образуется «брешь». Патогенные инфекционные агенты не встречают сопротивления и, «безнаказанно» заселив организм, развивают свою деятельность:
- деструктивную (разрушительную);
- токсическую.
причины
Гипогаммаглобулинемия может быть вызвана либо первичным или вторичным иммунодефицитом. Первичные иммунодефициты вызваны мутацией или ряда мутаций в геноме. Например, исследование , проведенное в 2012 году обнаружили , что соединение гетерозиготных безвредности мутации в гене CD21 связан с гипогаммаглобулинемией. Генетический анализ показал , пациент был гетерозиготным по CD21, с отечески унаследованным аллель (также совместно с одной сестрой) , имеющим нарушенным донорным сайтом сплайсинга в экзоне 6, в то время как наследуются по материнской линии аллель была мутация приводит в преждевременном стоп — кодоне в экзоне 13. Ни мутация была обнаружена в 100 здоровых контрольных субъектов, показывая редкость мутаций. Около 300 различных генов в общей сложности были идентифицированы , которые составляют различные формы первичного иммунодефицита (PID). Эти различные формы могут влиять на различные части иммунной системы, включая производство иммуноглобулина. Первичные иммунодефициты обычно имеют задержку в несколько лет между первоначальной клинической картиной и диагностикой. Некоторые первичные иммунодефициты включают атаксию-телеангиэктазию (AT), аутосомно — рецессивную АГАММАГЛОБУЛИНЕМИЮ (ARA), общие вариабельные иммунный (CVID), гипер-IgM синдромы , подкласс дефицит IgG, выделенные не-IgG недостатки иммуноглобулина, тяжелый комбинированный иммунодефицит (SCID), удельные дефицит антител (САД), синдром Вискотта-Олдрича , или болезнь брутона. CVID является наиболее распространенной формой первичного иммунодефицита. SCID считается неотложной медицинской помощи и предположительных случаев требуют немедленного специалиста центра направления для диагностики и лечения. Это чаще, чем гипогаммаглобулинемий развивается в результате другого условия, которые называются вторичными или приобретенным иммунодефицитом. Они включают рак крови , такие как хронический лимфоцитарный лейкоз (ХЛЛ), лимфомы или миеломы , ВИЧ , нефротический синдром , плохое питание, потерей белка энтеропатии , получая пересадку органа , или лучевой терапии . Это также включает в себя лекарственные препараты , которые могут вызвать гипогаммаглобулинемия , такие как кортикостероиды , химиотерапевтические препараты, или противосудорожные лекарства.
Диагностика болезни Брутона
Выявление агаммаглобулинемии происходить в несколько этапов:
-
Врач оценивает, как часто больной переносил различные инфекции за определенный промежуток времени.
-
Производится сбор семейного анамнеза: были ли случаи болезни Брутона у кого-то из мужчин. В случае положительного ответа больного срочно направляют на обследование к иммунологу.
-
Назначается лабораторный анализ крови на наличие защитных антител.
-
Проводится анализ на функционирование лимфоцитов, а также ряд генетически исследований.
-
При беременности можно провести пренатальную диагностику. В некоторых случаях дефекты иммунитета можно выявить у плода уже на ранних стадиях.
-









Профилактика
Данное заболевание является врожденным, а медицина еще не научилась предупреждать развитие таких патологий с помощью специфических методов. Следует ограничить влияние негативных факторов на организм, чтобы предупредить мутации генов, которые при передаче от родителей ребенку ведут к возникновению агаммаглобулинемии. Важными и действенными, хоть и не с 100% гарантией, являются абсолютно простые, доступные для соблюдения рекомендации:
- избегание радиоактивного облучения, замена диагностических и терапевтических радиоактивных методов на альтернативные;
- избегание воздействия температурных скачков, пребывание в зоне температурного комфорта;
- профилактика возникновения любых инфекционных заболеваний, а если они уже развились – их своевременное выявление и адекватное лечение;
- предупреждение, выявление и лечение эндокринных патологий, которые могут способствовать развитию агаммаглобулинемии;
- избегание воздействия любых агрессивных химических факторов на организм – бытовых, промышленных, сельскохозяйственных;
- проживание в экологически чистой местности;
- избегание воздействия профессиональных вредных факторов на организм, а если это невозможно – смена места работы;
- регулярное посещение с профилактической целью терапевта, который заподозрит первые признаки агаммаглобулинемии и далее скоординирует действия пациента для дальнейшего прохождения обследования и лечения;
- здоровый образ жизни в целом – отказ от любых вредных привычек, соблюдение правильного режима труда, отдыха, сна, питания, регулярные занятия физкультурой;
- закаливание.
О болезни
Болезнь Брутона, наследственная гипогаммаглобулинемия, агаммаглобулинемия – это определения одной и тоже болезни. Патология характеризуется:
- Врожденным происхождением;
- Мутациями в генотипе клеток;
- Недостаточностью синтеза В-лимфоцитов;
- Нарушением образования иммуноглобулина (их общее количество в крови снижается до нуля);
- Развитие на фоне снижения иммунитета воспалительных заболеваний ЛОР-органов, патологические новообразования в бронхах, в легких, болезни ЖКТ и с большими последствиями – воспаление мозга.
Чаще всего болезнь Брутона встречается у новорожденных мальчиков, нежели у девочек.
Диагностика
Признаки гнойно-воспалительных заболеваний, возникающих на фоне агаммаглобулинемии, известны. Но конкретный диагноз конкретной патологии ставят при инструментальном и лабораторном подтверждении. В диагностике учитывают анамнез (историю) заболевания.
При физикальном исследовании выявляются следующие нарушения в зависимости от локализации патологического процесса:
- при осмотре – отставание в физическом развитии, далее при взрослении и при затянувшемся течении нарушение общего состояния, бледность кожных покровов и видимых слизистых оболочек, сухость языка (он при этом покрыт белым налетом);
- при пальпации (прощупывании) – уменьшение и недоразвитость регионарных лимфатических узлов;
- при перкуссии (простукивании) – болезненность костных структур;
- при аускультации (прослушивании) – ослабление дыхания, хрипы.
Инструментальные методы исследования применяются в зависимости от локализации поражения. Это:
- рентгенография органов грудной клетки и брюшной полости;
- ультразвуковое исследование грудной клетки и брюшной полости;
- компьютерная томография (КТ) – является информативной при поражении костных структур;
- магнитно-резонансная томография (МРТ) – привлекается для изучения мягких тканей;
- биопсия костного мозга – забор фрагмента его тканей с дальнейшим изучением под микроскопом;
- биопсия лимфатических узлов.
и другие.
Из лабораторных методов исследования информативными являются:
- общий анализ крови – отмечается уменьшение количества B-лимфоцитов;
- иммунограмма – изучение количества иммуноглобулинов. Информативными являются определение IgA, IgM, IgG – их количество снижено либо они и вовсе отсутствуют;
- цитологическое исследование костного мозга – отсутствие в костном мозге плазмоцитов;
- гистологическое исследование – под микроскопом изучают лимфоидную ткань, отмечают в ее зародышевых центрах отсутствие плазматических клеток;
- бактериоскопическое исследование – изучают различные мазки в зависимости от пораженного органа (глотки, гортани, носовой полости, конъюнктивы и так далее), определяют в них возбудителя;
- бактериологическое исследование – делают посевы мазков на специальные питательные среды, через определенное время на них появляются колонии со специфическими признаками, по которым определяют инфекционного агента, присоединившегося на фоне сниженного иммунитета. С помощью данного метода также проводят определение чувствительности возбудителя к антибактериальным препаратам.
Диагностика
Первое – обратитесь к врачу иммунологу и аллергологу. Задача – выявить воспалительный процесс кожи, а также – подтвердить симптоматику снижения иммунитета.
Второе – полное обследование больного, подтверждающее отставание в физическом развитии.
Третье – прохождение лабораторного исследования, в частности, анализ крови на определение количества иммуноглобулинов – они могут быть снижены в сотни раз. При исследовании периферической крови обнаруживается дефицит В-клеток – их даже одного процента не будет диагностировано. При исследовании костного мозга – диагностировано отсутствие плазмоцитов.
Гистология лимфы показывает отсутствие клеток плазмы.
Для достоверности диагноза «наследственная агаммаглобулинемия» больной должен пройти диагностику вторичных нарушений иммунодефицита – ВИЧ, краснуху врожденного типа, токсоплазмоз и другие состояния.
Продукция антител
На рисунке 1 показаны этапы развития клеток и их взаимодействия, необходимые для продукции антител В-лимфоцитами. Лимфоциты происходят из стволовой клетки, общей для всех клеток гемопоэза. Ответвления в линию В-клеток происходит на этапе реаранжировки генов, кодирующих молекулы иммуноглобулинов. В основе разнообразия репертуара иммунной системы человека, с потенциальной возможностью продукции свыше 100 млн типов специфических антител, лежат несколько важных процессов:
- комбинационная изменчивость сочетание генов, кодирующих тяжелые и легкие цепи иммуноглобулинов;
- изменчивость связей генов;
- сочетание различных типов тяжелых и легких цепей;
- соматические мутации генов в процессе активации В-клеток.
Каждая молекула иммуноглобулина состоит из двух тяжелых и двух легких полипептидных цепей (рис. 2). В легких и тяжелых цепях можно выделить переменные или вариабельны (V) и постоянные части (С). В целом, постоянная часть молекулы влечет распределение иммуноглобулинов в организме и их биологические функции (взаимодействие с комплементом, секрецию в грудное молоко или секреты слизистых оболочек, возможность проходить через плаценту и тому подобное), тогда как переменная часть молекулы отвечает за связь с антигеном. Каждая В-клетка на поверхности имеет рецептор в виде уникального імуноглобулінового комплекса, что позволяет ей распознавать различные антигены.Для первичных В-клеток этот рецептор представлен антителами типа ІдМ. Связь антигена с імуноглобуліновим рецептором В-клетки активирует В-клетку к дальнейшему созреванию и дифференциации в конечную стадию – плазматичну клетку, способную продуцировать миллионы молекул иммуноглобулинов за короткий промежуток времени. Каждая В-клетка этого клона сохраняет антиген-специфическую реактивность первичной В-клетки. Некоторые В-клетки начинают продуцировать антитела различных типов, в которых одинакова последовательность антигенреактивної части молекулы сочетается с различными константными частями тяжелых цепей.Это обеспечивает продуцирование ІgG, ІдА и ІдЕ классов иммуноглобулинов с одинаковой антигенной специфичностью первичной В-клетки.
Симптомы агаммаглобулинемии
Какого-то единого проявления агаммаглоулинемии нет – клиническая симптоматика зависит от того, какие структуры организма были поражены. Как правило, это:
- органы дыхания;
- пищеварительная система;
- мозговые оболочки;
- орган зрения;
- ротовая полость;
- костные структуры;
- мышцы;
- кожа.
Наиболее часто агаммаглобулинемия приводит к развитию воспалительного поражения с образованием гноя.
Так как маленький человек рождается уже с врожденной патологией иммунной системы, он практически сразу становится беззащитным. Если мать кормит ребенка грудью, то барьером какое-то время являются защитные структуры, поступившие от нее с молоком. При этом четко прослеживается закономерность: отлучение от груди совпадает с началом различных заболеваний ребенка гнойно-воспалительного характера. Причем, одновременно могут быть поражены различные органы и системы, то ухудшает общее состояние пациента.







Следует заподозрить врожденную агаммаглобулинемию, если возникают и прогрессируют такие патологии, как:
- средний отит – поражение среднего уха;
- фронтит – воспаление слизистой оболочки, выстилающей лобную пазуху;
- бронхит – воспаление слизистой бронхов;
- трахеобронхит – сочетанное воспалительное поражение слизистой трахеи и бронхов;
- плеврит – поражение воспалительного характера, которое развивается в листках плевры;
- флегмона плевры – ее разлитое нагноение;
- энтерит – воспалительный процесс в слизистой тонкого кишечника;
- колит – такой же процесс со стороны толстого кишечника;
- энтероколит – сочетанное, одновременно развивающееся поражение слизистой большинства отделов тонкого и толстого кишечника;
- менингит – воспаление мозговых оболочек;
- конъюнктивит – воспаление слизистой оболочки глазного яблока;
- блефарит – поражение кожных покровов век;
- кератит – нарушение воспалительного характера в роговой оболочке глазного яблока;
- ирит – воспаление радужной оболочки;
- стоматит – воспалительный процесс, который распространяется в слизистой ротовой полости;
- остеомиелит – поражение костной ткани с образованием свищей (патологических ходов);
- миозит – воспаление мышц;
- фурункулез – гнойно-воспалительное поражение нескольких волосяных мешочков с окружающей тканью;
- пиодермия – поверхностное гнойное поражение кожных покровов;
- абсцесс – ограниченный гнойник.
Наиболее частые клинические проявления появляются при поражении:
- мозговых оболочек – головные боли, головокружение, нарушение сна;
- дыхательной системы – кашель с гнойным содержимым, одышка;
- пищеварительной системы – метеоризм, понос;
- органа зрения – затуманивание зрения, гнойные выделения из глаз;
- ротовой полости – мелкие гнойники;
- костных структур – боли, нарушение двигательной активности;
- мышц – боли в них;
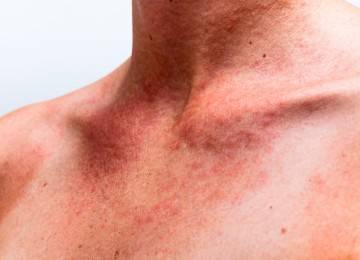
- кожи – различные виды сыпи.
Помимо местных, при агаммаглобулинемии выявляются нарушения общего состояния. Это:
- гипертермия – повышение температур тела. Может достигать 38,0-39,0 градусов по Цельсию;
- озноб;
- лихорадка – сочетанное появление гипетермии и озноба;
- общая слабость;
- разбитость;
- быстро возникающая утомляемость при прежних нагрузках;
- нарушение трудоспособности – физической и умственной;
- ухудшение сна – плохое засыпание, легкое пробуждение;
- ухудшение аппетита вплоть до его исчезновения;
- головные боли – нередко мигреневого характера.
эволюция соматических клеток и первичный иммунодефицит
В 2015 году журнал статью McDermott и др. сообщили о случае , в котором хромотрипсис , как правило , катастрофическое событие , в котором хромосома претерпевать массовое удаление и перестановку в ДНК единственного стебла клетки, вылечить пациент с синдромом Whim , первичным заболеванием иммунодефицита. КАПРИЗ является аутосомно — доминантным и обусловлен мутацией усиления из-функции от хемокин рецептора CXCR4. Мутация в CXCR4 увеличивает сигнализацию , поскольку она нарушает отрицательные элементы , обычно присутствующие регуляторные, создавая преувеличенные функции рецептора. Термин «КАПРИЗ» является аббревиатурой для основных проявлений заболевания: бородавок, гипогаммаглобулинемии, рецидивирующих инфекций и myelokathexis . Myelokathexis нарушается побег зрелых нейтрофилов из костного мозга, вызывая нейтропения . У больных с синдромом Whim серьезно уменьшить периферической крови В — клеток и некоторое снижение в периферической крови Т — клеток и моноцитов (McDermott). Вылечить пациента, обозначенное каприз-09, белая женщина имела в возрасте 58 лет , она также представлена с двумя дочерьми, КАПРИЗ-10 (возраст 21) и КАПРИЗ-11 (возраст 23). Обе дочери выполнены все клинические критерии синдрома прихоть, а КАПРИЗ-09 не сделал. Она сообщила , что у нее не было много серьезных инфекций , с детства до 38 лет , но не имел ни в течение последних 20 лет. Она не выполнила ни одного из критериев синдрома каприз за мягкой гипогаммаглобулинемия с тех пор , за исключением. КАПРИЗ-09 был первым пациентом никогда не описывался с myelokathexis, «М» при синдроме прихоти, и ее родители , братья и сестра не были никаких признаков синдрома. Таким образом, свидетельство совместимо с мутацией КАПРИЗА происходящего De Novo у пациента каприза-09, аутосомно — доминантный переход к двум из три ее дочерей, и спонтанная и полная ремиссия в капризе-09. Это обеспечивает первое доказательство того, что хромотрипсис может привести клинические преимущества, в частности, лечение генетического заболевания. Если клетка с хромотрипсисом умирает, это клинический незаметное, что делает истинная частоту его возникновения трудно определить. Таким образом, обнаруживается только тогда , когда оно приобретает сильное селективное преимущество создания клинически очевидной клональной популяции , несущей ту же схему делеций и механизмов. Это приводит либо рак, либо , если место является случайностью, излечение от генетического состояния , как это произошло у пациента каприза-09.
Причины агаммаглобулинемии

К развитию заболевания приводит геномная мутация
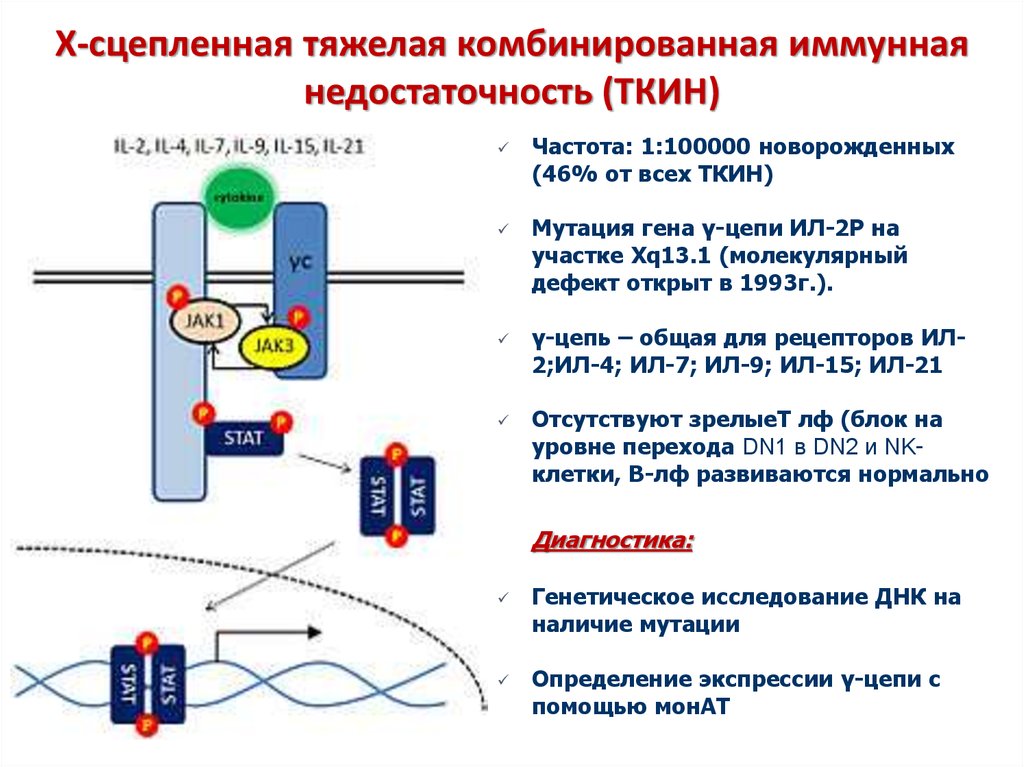
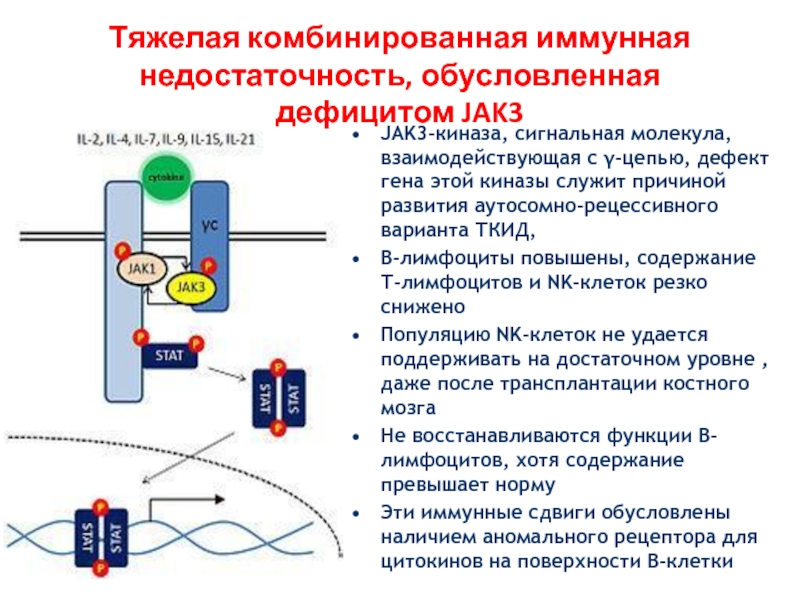
Только в конце 20 века была установлена главная причина заболевания — мутация гена, локализованного на Х-хромосоме. Этот ген содержит код фермента тирозинкиназы, необходимого для формирования предшественников В-лимфоцитов в красном костном мозге. Частота мутации в популяции составляет 1 случай на 200000 детей.
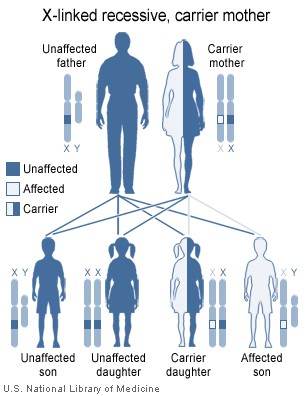
Так как тип наследования Х-сцепленный и рецессивный, заболевание проявляется только у мальчиков, имеющих, в отличие от девочек, всего одну Х-хромосому, поэтому мутация гена на этой хромосоме всегда имеет клиническое значение. Больные наследуют эту хромосому у матерей, являющихся гетерозиготными носительницами повреждённого гена. То есть одна из их Х-хромосом имеет нормальный генетический код, а вторая Х-хромосома — повреждённый. При этом клинически матери здоровы и не подозревают о носительстве дефектного гена.
На молекулярном уровне ген может быть повреждён в разной степени. Возможна замена нескольких нуклеотидов в цепочке ДНК, что приводит к прекращению синтеза необходимых аминокислот в составе тирозинкиназы и создаёт мало функциональный фермент. В этом случае возможна наработка минимального количества иммуноглобулинов в организме, но свои функции они смогут выполнять с трудом. Более массивные мутации (например, делеции) могут привести к полному прекращению синтеза тирозинкиназы и, соответственно, иммунных белков организма.
Симптомы и признаки агаммаглобулинемии
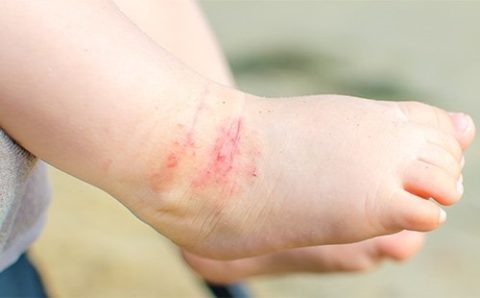
Начало проявлений заболевания у детей — 5-7 лет
Для этой патологии характерны повторные, длительно текущие, плохо поддающиеся лечению, тяжёлые инфекционно-воспалительные процессы в организме.
В первые 6-9 месяцев жизни в кровотоке новорождённого циркулируют иммуноглобулины, полученные от матери через сосуды плаценты и с грудным молоком (IgA, главным образом), поэтому явной клиники агаммаглобулинемии нет, наиболее яркие её проявления отмечают с двух до пяти-семи лет
В это время родители обращают внимание на частые инфекционные заболевания у ребёнка, вскоре принимающие характер хронического процесса, и начинают поиск причины
Общие симптомы: лихорадка, озноб, потливость, плохой аппетит, уменьшение массы тела, раздражительность, либо угнетение, сонливость.
Поражения кожи и подкожной клетчатки: пиодермия, экзема, фурункулы, карбункулы, абсцессы, флегмоны.
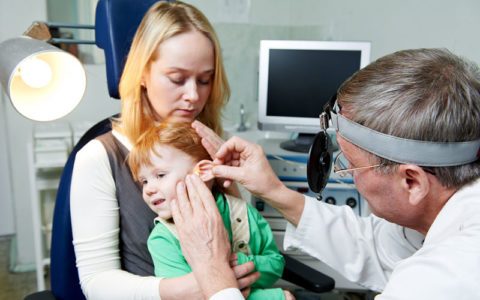
В патологический процесс могут вовлекаться любые органы
Поражения ротовой полости, глаз, органа слуха, пазух черепа: ангины, язвы слизистой рта, фарингиты, гнойные и вирусные конъюнктивиты, отиты, гаймориты, фронтиты и прочие синуситы вплоть до поражения всех пазух — пансинусита.
Поражения лёгких, сердца, кишечника: вирусные и бактериальные пневмонии, плевриты, миокардиты, бактериальные эндокардиты, энтероколиты, проктиты, парапроктиты.
Поражения костей: гнойный остеомиелит, артриты.
Критические состояния: кома, сепсис, дыхательная, сердечная, почечная недостаточность.
Лечение

Пациентам может понадобиться госпитализация
Лечением пациента занимается врач иммунолог и инфекционист.
Необходима пожизненная коррекция недостатка иммуноглобулинов путём введения в вену гамма-глобулинов или донорской плазмы.
Если заболевание диагностировано впервые, лечение проводится в режиме насыщения: гамма-глобулин вводится до общей массы его 3 г/л, но удобнее дозировать препарат по 200-400 мг на кг массы тела. Препарат вводят с интервалом 2-4 недели. Контроль уровня гамма-глобулинов, как правило, ведут по уровню IgG в сыворотке. После насыщения нужна пожизненная поддерживающая терапия профилактическими дозами препарата.


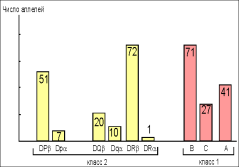
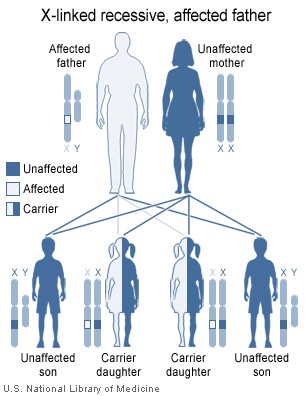

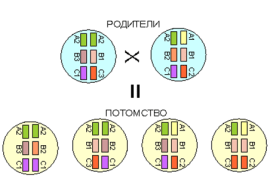


В случае инфекционного заболевания любой локализации необходима обязательная госпитализация пациента, повышение доз вводимых иммуноглобулинов, адекватная антибактериальная и противовирусная терапия. Наиболее современными антибиотиками считаются макролиды, фторхинолоны, новые поколения цефалоспоринов, иногда прибегают и к аминогликозидам, всё реже — к антибиотикам пенициллинового ряда.
В зависимости от локализации процесса используются препараты для поддержки повреждённого органа, антисептики, инфузионная терапия.
Клиническая картина
При рождении ребенка заболевание клинически не проявляется. Содержание иммуноглобулинов в это время обычно нормальное за счет пассивно полученных трансплацентарно материнских Однако отсутствие IgM и IgA у ребенка в возрасте одного месяца может вызвать подозрение на наличие АГАММАГЛОБУЛИНЕМИИ в тех случаях, когда имеется неблагоприятный семейный анамнез (внезапная смерть братьев или сестер больного в первые месяцы после рождения).
Повышенная чувствительность к инфекции выявляется с 3—5-месячного возраста, что клинически проявляется стойкой пиодермией, сепсисом, которые плохо поддаются антибактериальной терапии и склонны к прогрессированию. В дальнейшем септический процесс распространяется на легкие (тяжелая пневмония с абсцедированием, бронхоэктазы), среднее ухо (хронический гнойный средний отит, антрит), желудочно-кишечный тракт (рецидивирующий язвенный энтероколит). Возможны также менингит, пансинусит, остеомиелит.
Нередким является спруподобный синдром. Отмечено обычное течение туберкулеза и вирусных инфекций (кори, полиомиелита, эпидемического паротита, ветряной оспы), что указывает на связь гуморального иммунитета к этим заболеваниям с другими белковыми фракциями, с компенсаторным влиянием системы пропердина, которая у больных АГАММАГЛОБУЛИНЕМИЕЙ функционирует нормально.
Комбинированные формы АГАММАГЛОБУЛИНЕМИИ, в отличие от изолированных, характеризуются меньшей устойчивостью к вирусным инфекциям, сопровождаются гипоплазией всех элементов лимфатической системы.