Клиническая картина
Симптомы фенилкетонурии проявляются не сразу после рождения, а по мере накопления в организме фенилаланина и продуктов его метаболизма. Чаще всего первые проявления заболевания развиваются в возрасте 2 месяцев, при отсутствии лечения к 6 месяцам картина становится более отчетливой.
Первыми тревожными сигналами становятся вялость и апатия у малыша, хотя ребенок и наоборот может быть тревожным, легко возбудимым, возможны эпизоды рвоты. Кроме того со временем могут присоединяться судорожные припадки по типу эпилептиформных, наблюдаться гипертонус мышц, гиперкинезы, тремор конечностей, атаксия. Также специфическим признаком является своеобразный «мышиный» запах, исходящий от ребенка.
К 6-ти месяцам становится очевидным отставание в психическом развитии, вплоть до развития олигофрении и идиотии.
В физическом развитии отставание не столь явное, но тоже есть. Оно выражается уменьшением размера черепа, более поздним прорезыванием зубов, нарушением ползания и хождения. Походка у них тоже своеобразная: дети ходят мелкими шажками, раскачиваясь, а стоят с широко раздвинутыми ногами, согнутыми в тазобедренных и коленных суставах.
Основные причины болезни
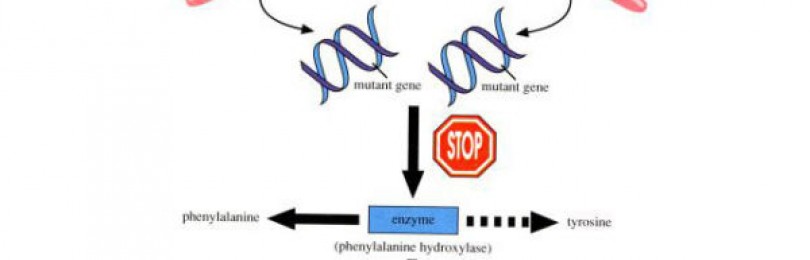
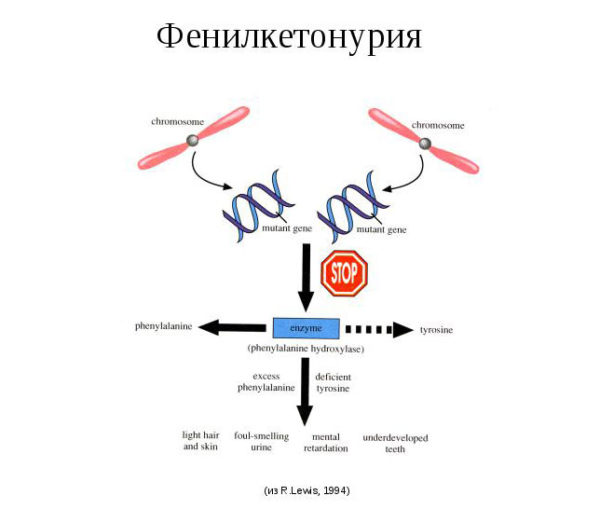
Основная причина развития болезни – наследование детьми от обоих биологических родителей мутантного гена. Таким образом, заболевание считается аутосомно-рецессивным.
В большинстве случаев возникает вследствие мутационных изменений гена, находящегося на длинном плече 12 хромосомы и отвечающего за кодирование фермента фенилаланин-4-гидроксилазы.
Такой вариант болезни является классической ФКУ и диагностируется у 98 больных детей из ста.
Гиперфенилаланинемия у больных при обследовании может доходить до 30 мг% и больше. Отсутствие своевременной терапии становится причиной умственной отсталости, и чаще всего она выражена значительно.
При фенилкетонурии II типа генетический дефект находится в 4-й хромосоме, прогноз неблагоприятный – смерть малыша вследствие необратимых изменений обычно происходит на 3 году жизни.
При III типе заболевания мутация находится в 11 хромосоме, эффективного лечения нет, поэтому при данном диагнозе развивается тяжелая умственная отсталость.
Считается что риск рождения ребенка у пар – гетерозиготных носителей дефектного гена возрастает в следующих случаях:
- При хроническом алкоголизме отца или матери;
- Если в период беременности у женщины и ее партнера были инфекционно-воспалительные заболевания половых органов;
- При негативном влиянии на организм родителей плохих экологических условий, вредной работы, хронических болезней.
Фенилкетонурия – симптомы
При рождении ребенок с данным диагнозом выглядит здоровым, и только спустя 2-6 месяцев обнаруживаются первые симптомы. Фенилкетонурия признаки начинает проявлять тогда, когда в детском организме накапливается фенилаланин, поступающий вместе с грудным молоком или смесями для искусственного вскармливания. Могут отмечаться такие, пока еще не специфические симптомы:
- чрезмерная вялость либо беспокойство;
- беспричинные крики;
- мышечная дистония;
- частые срыгивания;
- судороги;
- нарушения сна.
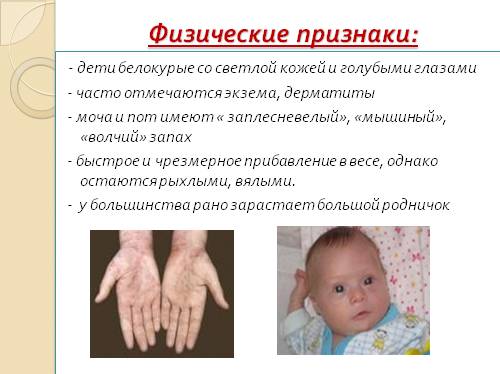
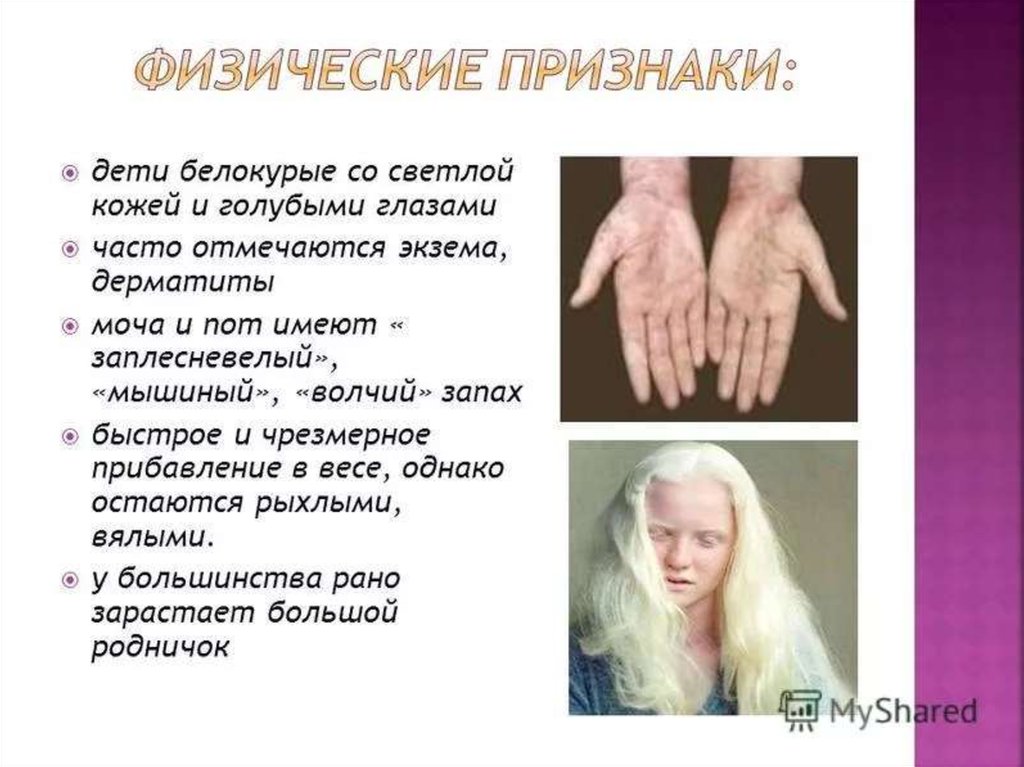
Помимо этого, больные малыши имеют более светлый кожный покров, волосы и глаза, чем у здоровых членов семьи, что связано с нарушением выработки в организме пигмента меланина. Еще один диагностический признак, который могут заметить медики или внимательные родители, – своеобразный «мышиный» запах, вызываемый выделением фениланина с мочой и потом.
Более явными клинические проявления становятся примерно в полугодовалом возрасте, после введения первого прикорма:
- неспособность фокусирования взгляда на отдельных предметах;
- безучастность в отношении всего происходящего;
- отсутствие мимики, улыбки;
- тремор рук и так далее.
Заметны и физические отклонения: маленький размер головы, выдающаяся вперед верхняя челюсть, отставание в росте. Больные детки поздно начинают держать голову, ползать, садиться, вставать. Характерна особая поза в положении сидя – поза «портного», при которой ручки постоянно согнуты в локтях, а ножки – в коленях. В возрасте трех лет, если лечение так и не начато, симптоматика нарастает.
Диагностика
Фенилкетонурия является областью исследования генетиков, психиатров, невропатологов и гепатологов. Окончательный диагноз ставят на основании следующих данных:
- обнаружение в крови фенилаланина в количестве более 900 мгмоль/л;
- положительный результат пробы Феллинга;
- присутствие в крови фенилмолочной, фенилуксусной или фенилпировиноградной кислоты.
Важными критериями диагностики фенилкетонурии у детей являются жалобы родителей на нетипичное поведение, задержку психофизического развития ребенка, прочие частые при ФЛК симптомы.
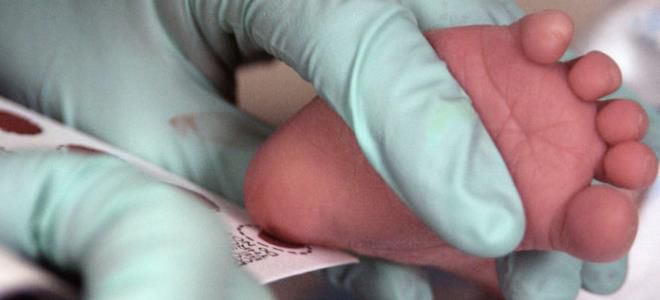
Высокой достоверностью обладает ранняя диагностика заболевания, еще в период внутриутробного развития плода. Сегодня каждая женщина, наблюдающаяся у гинеколога по поводу беременности, проходит ряд скрининговых обследований, в том числе и на фенилкетонурию.






Диета для детей дошкольного возраста и школьников
По мере адаптации организма к фенилаланину детям с возраста 5 лет можно постепенно уменьшать ограничения в питании. Расширение рациона происходит путем введения круп, молочных продуктов, мясных изделий. Школьники старших классов имеют уже высокую толерантность к фенилаланину, поэтому в этом возрасте можно продолжить расширение диеты, при этом необходимо отслеживать реакцию на все изменения в питании. Для контроля состояния ребенка применяются следующие способы:
- оценка неврологических показателей, психологического состояния;
- контроль показателей электроэнцефалограммы;
- определение уровня фенилаланина.
Механизм развития заболевания
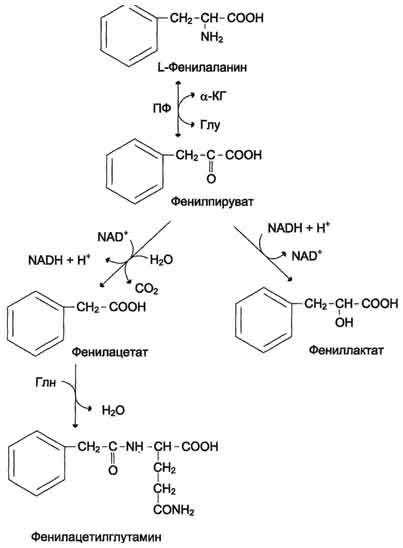
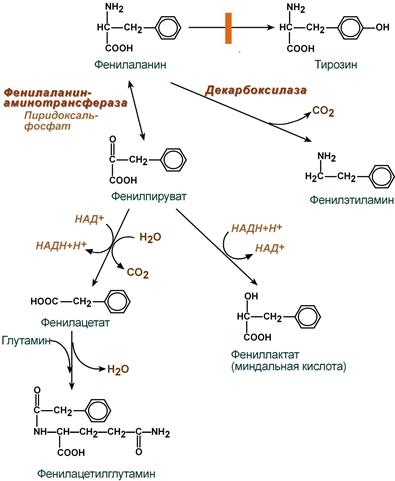
Причинообразующим фактором возникновения генных нарушений является метаболический блок, который препятствует образованию фенилаланин-4-гидроксилазы (фермент, отвечающий за превращение аминокислоты фенилаланина в тирозин). Протеиногенная аминокислота тирозин является составной частью белков и пигмента меланина, поэтому является необходимым элементом для функционирования всех систем организма, а ее нехватка приводит к ферментопатии.
Следствием подавления образования метаболита, вызванного мутационной инактивацией фермента, является активирование вспомогательных путей обмена фенилаланина. Ароматическая альфа-аминокислота в результате дефектных обменных процессов распадается на токсические производные, которые в нормальных условиях не образуются:
- фенилпировиноградную кислоту (фенилпируват) – жирно-ароматическая альфа-кетокислота, ее образование приводит к миелинизации отростков нейронов и слабоумию;
- фенилмолочную кислоту – продукт, образовавшийся при восстановлении фенилпировиноградной кислоты;
- фенилэтиламин – начальное соединение для биологически активных передатчиков электрохимических импульсов, повышает концентрацию дофамина, адреналина и норадреналина;
- ортофенилацетат – токсичное вещество, вызывающее нарушение обменных процессов жироподобных соединений в головном мозге.
Медицинская статистика свидетельствует о том, что патологически измененный ген присутствует у 2% населения, но при этом он никак не проявляет себя. Генетический дефект передается ребенку от родителей только при наличии заболевания у обоих партнеров, при этом младенец в 50 % случаев становится носителем мутировавшего гена, оставаясь здоровым. Вероятность того, что фенилкетонурия у новорожденных приведет к заболеванию, составляет 25%.
По какому типу наследуется
Болезнь Феллинга является генетическим отклонением, наследуемым по аутосомно-рецессивному типу. Такой тип наследования означает, что развитие признаков врожденного заболевания произойдет только при наследовании ребенком по одной дефектной генокопии от обоих родителей, которые являются гетерозиготными носителями видоизмененного гена.
Развитие врожденного заболевания в 99% случаев вызывает мутация гена, ответственного за кодирование фермента, обеспечивающего синтез фенилаланин-4-гидроксилазы (классическая фенилкетонурия). До 1% генетических заболеваний связаны с мутационными изменениями, происходящими в других генах, и вызывающих недостаточность дигидроптеридинредуктазы (ФКУ II типа) или тетрагидробиоптерина (ФКУ III типа).
Симптомы
 У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина. Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса. Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей. Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами. Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах. Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
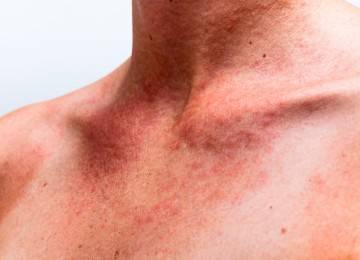
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
Фенилкетонурия – что это за болезнь?
Фенилкетонурия, или болезнь Феллинга, является тяжелой патологией, впервые описанной в 1934 году норвежским ученым Феллингом. Тогда Феллинг провел обследование нескольких детей с умственной отсталостью и выявил у них присутствие в моче фенилпирувата – продукта распада поступающей с едой аминокислоты фениланина, которая в организме больных не расщепляется. Фенилкетонурия – заболевание, связанное с нарушением обмена веществ врожденного характера, открытое одним из первых.







Фенилкетонурия – тип наследования
Болезнь Феллинга является хромосомно-генетической, наследственной, передаваемой детям от родителей. Виновником развития патологии выступает ген, находящийся на 12 хромосоме. Он ответственен за производство печеночного фермента фенилаланин-4-гидроксилазы, за счет которого происходит превращение фенилаланина в другое вещество – тирозин (оно требуется для нормальной работы организма).
Установлено, что фенилкетонурия наследуется как рецессивный признак. Приблизительно 2 % людей являются носителями дефектного гена, но при этом не страдают фенилкетонурией. Патология развивает только тогда, когда и мать, и отец передают ген ребенку, а случиться это может с вероятностью 25 %. Если фенилкетонурия наследуется как рецессивный признак, жена гетерозиготна, а муж гомозиготен по нормальному аллелю гена, то вероятность того, что дети будут здоровыми носителями гена фенилкетонурии, равна 50 %.
Формы фенилкетонурии
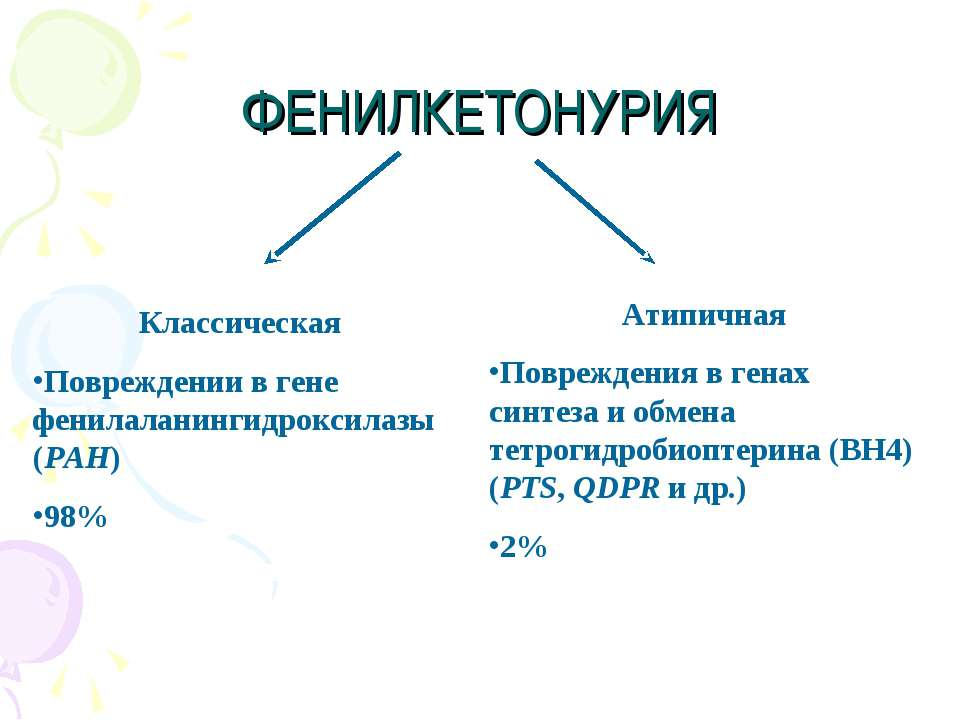
Рассматривая, у кого может развиться фенилкетонурия, что это за заболевание, зачастую речь ведется о классической форме патологии, которая встречается примерно в 98 % случаев. Остальные случаи – кофакторная фенилкетонурия, обусловленная дефектом тетрагидробиоптерина вследствие нарушения его синтеза или восстановления активной формы. Данное вещество служит кофактором ряда ферментов, и без него невозможно проявление их активности.

Фенилкетонурия – причины
Болезнь Феллинга является патологией, при которой из-за мутаций в гене, вызывающих дефицит или отсутствие фенилаланин-4-гидроксилазы, происходит накопление в тканях и физиологических жидкостях фенилаланина, а также продуктов его неполного расщепления. Часть избыточного фенилаланина превращается в фенилкетоны, выводимые с мочой, что и обусловило название болезни.
Нарушение обменных процессов сказывается в большей степени на головном мозге. На его ткани производится отравляющее воздействие, нарушаются процессы жирового обмена, происходит сбой миелинизации нервных волокон, снижение образования нейромедиаторов. Так начинается запуск патогенетических механизмов задержки умственного развития у ребенка.
Прогноз и профилактика
Предупредить развитие необратимых повреждений органов ЦНС позволяет ранняя диетотерапия. Для того чтобы лечебное питание было назначено вовремя детям практически сразу после рождения проводится массовый скрининг и при необходимости назначается дополнительная диагностика.
Своевременное проведение элиминационной диеты и неукоснительное соблюдение предписаний врача по лечебному питанию позволяет родителям вырастить полностью здорового ребенка, то есть без отклонений в интеллектуальном, физическом и психическом развитии.
Прогноз течения патологии неблагоприятный при поздно начатой диетотерапии. Если в подростковом возрасте диета расширяется неадекватно или ее соблюдение полностью прекращается, то наблюдается снижение способности к обучаемости, нарушение поведенческих норм, расстройства со стороны психики.
Вот почему большинство психотерапевтов рекомендуют придерживаться элиминационной диеты как минимум до 18 лет.
Риск рождения детей, больных фенилкетонурией, оценивается после обследования супружеских пар в медико-генетических центрах. Обязательно такому обследованию должны подвергаться супруги, у которых уже рожден ребенок с таким заболеванием.
Желательно чтобы при планировании зачатия обследование на генетические патологии проходили пары, имеющие близкое кровное родство.
При атипичных формах ФКУ диетотерапия нужных результатов коррекции расщепления фенилаланина не дает. Прогноз течения таких типов патологии неутешительный – малыши либо гибнут в первые годы рождения, либо у них наблюдается глубокая умственная отсталость.
Фенилкетонурия пока единственное наследственное заболевание, раннее лечение которого предупреждает практически на 100 процентов вероятность развития тяжелых осложнений в будущем.
Поэтому родителям нельзя отказываться от неонатального скрининга их малыша в роддоме, а при рождении ребенка дома нужно его обследовать в течение первых трех недель жизни.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин
Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека. Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются
Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ. Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты. Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.






Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
 Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие. В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная). После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся к содержанию фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
 Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Патогенез
При фенилкетонурии в печени больного не продуцируется особый фермент под названием фенилаланин-4-гидроксилаза.
Основная его функция – преобразование поступающего в органы ЖКТ с пищей фенилаланина в аминокислоту тирозин. Это аминокислота входит в состав большинства ферментов, гормонов, способствует выработке меланина (пигмент), принимает участие в образовании белков и в функционировании большинства внутренних органов.
Метаболический блок при фенилкетонурии неуклонно приводит к тому, что начинают работать побочные (атипичные) пути обмена фенилаланина, в результате он трансформируется в те вещества, которых в теле здорового человека быть не должно.
Это кислоты – фенилмолочная и фенилпировиноградная, ортофенилацетат, фенилэтиламин. По сути, они являются токсическими соединениями, и их накапливание в кровеносной системе приводят:
- К нарушению нормального обмена липидов (жировых клеток) в разных отделах головного мозга;
- К нехватке нейромедиаторов, ответственных за бесперебойную передачу нервных импульсов во всей нервной системе.
В результате это приводит к прогрессирующему снижению интеллекта и к умственной отсталости выраженной степени – олигофрении, имбецильности, идиотии.
Отсутствие терапии становится причиной того, что через каждые 10 недель коэффициент развития интеллекта больного малыша падает на 5 пунктов.
Ответы на часто задаваемые вопросы
Как проявляется фенилкетонурия у новорожденных?
Как выглядят больные фенилкетонурией?
- посветление волос и радужки глаза из-за недостатка пигмента меланина
- чрезмерная прибавка в весе
- быстро зарастает большой родничок
- суховатая кожа
- шелушение, сыпь и экзема
- частая рвота
- моча и пот с характерным «мышиным» запахом
- появляются судороги и спазмы
- скованность движений и зажатая «поза портного», что связанно с повышенным напряжением в мышцах
- неадекватное поведение, выкрики, смех
- уменьшение размеров черепа
- деформация ушных раковин
- дрожание пальцев рук
- недержание мочи
- выступающая вперед нижняя челюсть
Какие смеси использовать для ребенка с фенилкетонурией?
витаминымикроэлементыДля детей до одного года рекомендуют:
- Афенилак 13, Афенилак 15 от компании «Нутритек», Россия;
- MIDмил ФКУ 0 (Hero, Испания);
- ХР Аналог («Нутриция», Голландия);
- Фенил Фри 1 («Мид Джонсон» США).
Для детей старше одного года и для взрослых:
- П-АМ 1, П-АМ 2, П-АМ 3;
- Изифен (готовый продукт), а также ХР Максамейд и ХР Максамум с нейтральным и фруктовым вкусами («Нутриция», Голландия).
Какие бывают типы фенилкетонурии?
фенилкетонурии
- Фенилкетонурия I. Классическая и наиболее распространенная форма заболевания, описанная выше в статье. Связана с мутацией гена в 12-й хромосоме, при этом нарушается образование фермента фенилаланин-4-гидроксилазы, который превращает фенилаланин в тирозин.
- Фенилкетонурия II. При этой форме заболевания нарушение происходит в 4-й хромосоме. Нарушается выработка фермента дигидроптеридинредуктазы, который также способствует превращению фенилаланина в тирозин. Заболевание наследуется так же, как и I форма: для того, чтобы родился больной ребенок, необходимо, чтобы носителями гена были оба родителя. Распространенность фенилкетонурии II – 1 случай на 100 000 новорожденных.
- Фенилкетонурия III. В результате генетических нарушений возникает недостаток фермента 6-пирувоилтетрагидроптеринсинтазы. Наследуется, как и две предыдущие формы заболевания. Распространенность – 1 случай на 300 000 новорожденных.
Дают ли инвалидность при фенилкетонурии?
Критерии установления инвалидности при фенилкетонурии
- При фенилкетонурии I инвалидность устанавливают только при необратимых нарушениях со стороны центральной нервной системы, которые приводят к неврологическим расстройствам и умственной отсталости.
- При фенилкетонурии II и III типа группу инвалидности устанавливают во всех случаях.
Существует ли профилактика фенилкетонурии?
- Генетическое консультирование. Необходимо людям, планирующим завести ребенка, которые больны или являются носителями неправильного гена, у которых болен хотя бы один близкий родственник или уже родился больной ребенок. Консультирование проводит врач-генетик. Он помогает разобраться, как ген, ответственный за фенилкетонурию, передавался в предыдущих поколениях, каковы риски будущего ребенка. Также генетик помогает с планированием семьи.
- Скрининг новорожденных. Анализ не помогает предотвратить заболевание, но позволяет выявить его максимально рано, пока оно еще не привело к необратимым изменениям в головном мозге.
- Консультации и диета для женщин, страдающих фенилкетонурией. Если вы женщина и страдаете ФКУ, вам следует проконсультироваться с врачом и спросить, когда лучше планировать беременность в вашем случае. Во время беременности нужно соблюдать правильную диету – это помогает предотвратить дефекты развития у ребенка.
Каковы факторы риска фенилкетонурии?
- Как уже упоминалось в статье, ребенок рискует получить заболевание или стать носителем мутантного гена, если он есть у обоих родителей.
- Среди разных этнических групп распространенность фенилкетонурии различается. Например, среди представителей негроидной расы неправильный ген встречается реже.
- В группе повышенного риска находятся дети матерей, страдающих фенилкетонурией. Если во время беременности женщина не придерживается специальной диеты, у ребенка могут возникать дефекты развития.
Описание патологии
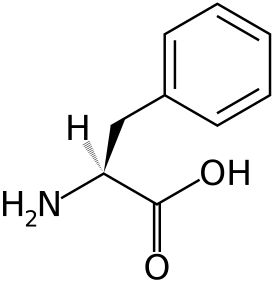
Развитие фенилкетонурии (ФКУ) связано с неспособностью организма человека с дефектным геном расщеплять поступающую вместе с пищей, богатой на белки, аминокислоту под названием фенилаланин.
В итоге кислота и элементы ее распада скапливаются в тканях и биожидкостях, превращаются в токсические соединения, под воздействием которых происходит глубокое и в большинстве случаев необратимое поражение органов ЦНС.
Отсутствие своевременного адекватного лечения становится причиной задержки развития интеллекта вплоть до олигофрении.
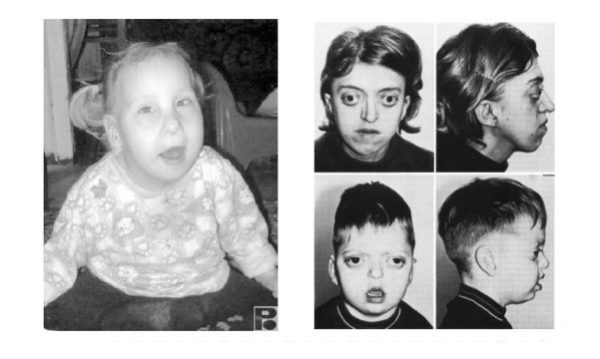
Манифестирует Фенилкетонурия обычно у больных малышей в первые 3-6 месяцев после рождения.
Выявление заболевания до появления первых клинических проявлений и соблюдение в дальнейшем эллиминационной диеты исключает развитие тяжелых, необратимых расстройств нервной системы у детей и взрослых, то есть опасности для физического, психоэмоционального и умственного здоровья нет.
Фенилкетонурия в среднем диагностируется у 1 новорожденного из 10 тысяч. В Турции количество больных большое — детей с ФКУ рождается 1 на 2, 5 тысячи, в Японии – 1 на 100 тысяч рожденных, в Африке (у представителей негроидной расы) болезнь встречается исключительно редко. У девочек патология диагностируется в 2 раза чаще по сравнению с мальчиками.

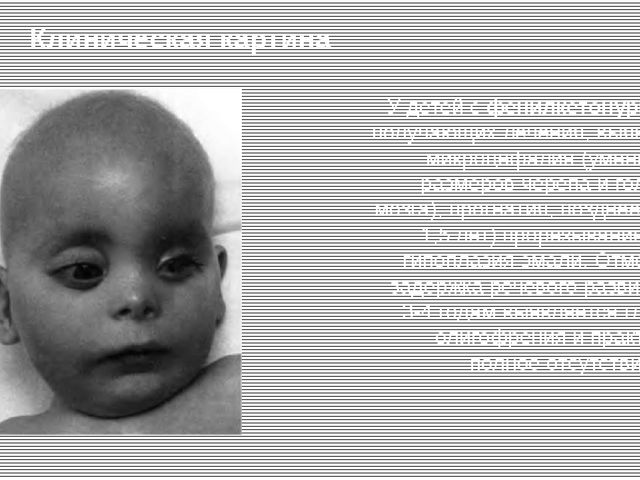




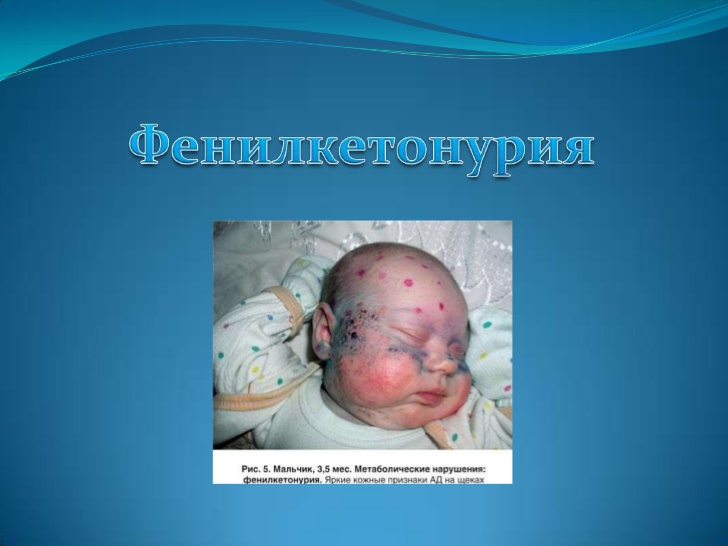
В медицинской литературе можно встретить и другие обозначения болезни — фенилпировиноградная олигофрения, болезнь Феллинга.
Группы продуктов при ФКУ
В рацион питания пациентов с ФКУ наряду с малобелковыми крахмалистыми продуктами и лечебными смесями входят и продукты натурального происхождения. При составлении меню следует четко рассчитывать количество потребляемого белка и не превышать рекомендованную врачом дозировку. Для исключения токсического влияния на организм разработаны 3 списка продуктов, которые содержат запрещенные (красный), нерекомендованные (оранжевый) и разрешенные (зеленый) позиции.
Красный список
Фенилкетонурия развивается на фоне отсутствия фермента, превращающего фенилаланин в тирозин, поэтому высокое содержание белка является поводом для отнесения продуктов в запрещенный (красный) список. Позиции из этого перечня следует полностью исключить рациона питания больного ФКУ:
- мясо;
- внутренние органы животных, субпродукты;
- колбасы, сосиски;
- морепродукты (в т.ч. рыба);
- яйца всех птиц;
- кисломолочные продукты;
- орехи;
- плоды бобовых и зерновых культур;
- соевые продукты;
- желатинсодержащие блюда;
- кондитерские изделия;
- аспартам.
Оранжевый список
Продукты, которые должны дозировано поступать в организм ребенка с диагнозом ФКУ, включены в оранжевый список. Включение в рацион питания позиций из этого перечня допустимо, но в строго ограниченном количестве. Эти продукты хоть и содержат не много белка, но тоже могу повысить уровень фенилаланина, поэтому их употребление не рекомендовано:
- консервированные овощи;
- блюда из картофеля и риса;
- капуста;
- молоко;
- щербет.
Зеленый список
Безбелковые продукты разрешены к употреблению больными с диагнозом фенилкетонурия без ограничений. Перед покупкой позиций из зеленого списка необходимо изучить состав, указанный на упаковке, и убедиться, что там не содержится краситель аспартам, содержащий фенилаланин:
- фрукты;
- овощи (за исключением картофеля и капусты);
- ягоды;
- зелень;
- крахмалистые крупы (саго);
- мед, сахар, варенье;
- мучные изделия из кукурузной или рисовой муки;
- масла, жиры (сливочное, растительное, оливковое).
Виды
Заболевание может протекать по классическому и атипичному варианту. К атипичным формам болезни относят:
- ФЕНИЛКЕТОНУРИЮ II ТИПА. Мутация гена приводит к дефициту дигидробиоптерин-редуктазы, в результате активность соединения, ответственного за преобразование фенилаланина, нарушается. Одновременно в сыворотке крови и в цереброспинальной жидкости больного выявляется дефицит витамина В9, от нормального количества которого зависит утилизация (расщепление и выведение) аминокислот.
- ФЕНИЛКЕТОНУРИЮ III ТИПА. Развивается вследствие нехватки катализатора, необходимого для выработки тетрагидробиоптерина (преобразует поступающий в организм фенилаланин в тирозин).
- ПРИМАПТЕРИНУРИЮ. Выявляется при незначительной степени гиперфенилаланинемии. До настоящего времени ферментная мутация данного вида фенилкетонурии еще не установлена. Но у больных в моче обнаруживается избыточное количество примаптерина и его соединений, а в спинномозговой жидкости уровень нейромедиаторных метаболитов остается в пределах нормы.
Атипичные типы ФКУ сходны по клиническим симптомам с классическим течением патологии, но при их развитии даже своевременное лечение и диетотерапия не останавливают необратимые изменения в функционировании органов ЦНС.
Отдельно выделяют материнскую фенилкетонурию, заболевание диагностируется у детей, рожденных женщинами с ФКУ, которые не соблюдали лечебное питание.
Повышенный уровень фенилаланина вызывает ряд врожденных аномалий развития головного мозга:
- Вентрикуломегалию – увеличение желудочков головного мозга в размерах;
- Задержку миелинизации (формирование оболочек нервных клеток) и гипоплазию (недостаточное развитие) белого вещества мозга;
- Низкий вес головного мозга по сравнению с нормой.
Материнская фенилкетонурия становится причиной хронической интоксикации развивающегося плода токсическими соединениями, и как следствие этого приводит к задержке развития умственной функции рожденных детей.