Дегенеративные изменения
Разрушение нейрона наступает по нескольким причинам:
- Накопление внутри клетки патологических белков, вызывающих ее гибель;
- Перестройка протеинов инициируемое прионом;
- Наследственные изменения ферментных или белковых систем.
При таупатиях происходит нарушение работы протеина клетки. Этот белок фиксирует микротрубочки. Эти структуры выполняют опорную функцию и передачу питательных веществ. При изменении структуры таубелка микротрубочки перестают функционировать, а клетка гибнет. К таупатиям относят болезнь Альйгеймера, Пика, фронтотемпоральную дегенерацию.
Синуклеопатии характеризуются прогрессирующим накоплением в тканях альфа-амилоида. Например, болезнь Паркинсона или деменция с тельцами Леви. Ученые предполагают, что клетки разрушаются именно от накоплений эти белковых гранул. Но последние разработки препаратов показывают, что избавление от них не приводит к излечению больного. Возможно, что белок внутри клетки появляется вторично и не участвует в процессе ее гибели.
Патогенез прионных болезней более понятен. Прион – это небольшой белок, который заставляет клетку вырабатывать себе подобные протеины. Эти вторичные молекулы полностью напоминают его структуру и не могут использоваться в организме. Мозговое вещество истощается. А его структура напоминает губку. Конечно, интеллектуальные и контролирующие функции ЦНС утрачиваются.
Прогрессирующее нейродегенеративное заболевание
На сегодня только несколько дегенеративных болезней научились лечить. В основном их развитие идет по нарастающей. Вначале недуга патология легкая или мало заметная. А в конце формируется тяжелая инвалидность.

Прогрессирующее течение у болезни Паркинсона, Альцгеймера, деменции с тельцами Леви, патологии Пика. Врачам удается контролировать рассеянный склероз и спинальную мышечную атрофию. К сожалению, последствия такой терапии непредсказуемы. Пациентам вводят довольно агрессивные препараты. И неизвестно, как это повлияет на их здоровье.
Симптомы синдрома Лея
Характерные для синдрома Лея симптомы выглядят следующим образом:
- тошнота, рвота;
- быстрое снижение массы тела;
- ухудшение аппетита;
- задержка психомоторного развития;
- тонико-клонические судороги;
- приступы мышечной дистонии, гипотонии;
- респираторные аномалии;
- тремор конечностей;
- нарушение координации;
- нарушение акта глотания;
- атрофия зрительных нервов (вплоть до слепоты);
- нарушение сухожильных рефлексов;
- нарушения сознания;
- быстрая утомляемость;
- сонливость.
Если Вы обнаружили у себя схожие симптомы, незамедлительно обратитесь к врачу. Легче предупредить болезнь, чем бороться с последствиями.
Лучшие врачи по лечению синдрома Лея
10
Рефлексотерапевт
Невролог
Мануальный терапевт
Врач высшей категории
Шнигирист Александр Ильич
Стаж 28
лет
Кандидат медицинских наук
Евромедклиник
г. Москва, Сиреневый бульвар, д. 32а
Щелковская
900 м
Первомайская
980 м
8 (495) 185-01-01
9
Вертебролог
Рефлексотерапевт
Невролог
Мануальный терапевт
Врач высшей категории
Пятков Сергей Анатольевич
Стаж 21
год
Кандидат медицинских наук
Евромедклиник 24 Жулебино
г. Москва, Люберцы, м-н Городок Б, ул. 3-е Почтовое Отделение, д. 102
Жулебино
830 м
Котельники
950 м
8 (499) 969-25-84
8.9
Невролог
Врач второй категории
Аносова Мария Олеговна
Стаж 14
лет
Медцентр Столица на Арбате
г. Москва, Большой Власьевский пер., д. 9
Смоленская
760 м
Кропоткинская
1 км
Смоленская
1.3 км
Медцентр Столица на Бабушкинской
г. Москва, ул. Летчика Бабушкина, д. 48Б
Бабушкинская
1.5 км
Медстайл эффект на Достоевской
г. Москва, 3-й Самотёчный пер., д. 2
Достоевская
330 м
Новослободская
990 м
Цветной бульвар
1 км
8 (499) 519-39-10
8 (499) 519-36-01
8 (499) 519-39-98
8.3
Вертебролог
Кинезиолог
Гериатр (геронтолог)
Невролог
Вегетолог
Реабилитолог
Рефлексотерапевт
Врач первой категории
Кубышта Светлана Михайловна
Стаж 15
лет
Ист клиник в Беляево
г. Москва, Профсоюзная, д. 104
Беляево
110 м
Коньково
890 м
Калужская
1.8 км
Альфа-Центр Здоровья
г. Москва, Комсомольский пр-т, д. 17, стр. 11
Фрунзенская
480 м
Парк культуры
1.1 км
Парк культуры
1.3 км








8 (495) 185-01-01
8 (499) 519-36-58
8.9
Невролог
Мануальный терапевт
Врач первой категории
Бабенко Михаил Федорович
Стаж 16
лет
ABC медицина на 1905 года
г. Москва, ул. 1905 года, д. 17
Улица 1905 года
290 м
Краснопресненская
1.4 км
8 (499) 519-36-05
9.8
Невролог
Гадаборшева Тамара Магомедовна
Стаж 42
года
Медцентр Медквадрат на Каширском ш.
г. Москва, Каширское ш., д. 74, стр. 1
Каширская
2.8 км
Каширская
3 км
8 (499) 519-35-25
9.9
Невролог
Эпилептолог
Врач высшей категории
Романова Анна Вячеславовна
Стаж 8
лет
Кандидат медицинских наук
Медцентр Медквадрат на Воротынской
г. Москва, ул. Воротынская, д. 4
Планерная
4.6 км
Медцентр Медквадрат на Каширском ш.
г. Москва, Каширское ш., д. 74, стр. 1
Каширская
2.8 км
Каширская
3 км
8 (499) 519-35-25
8 (499) 519-35-25
10
Рефлексотерапевт
Гирудотерапевт
Ревматолог
Пульмонолог
Невролог
Кардиолог
Врач высшей категории
Сафиуллина Аделия Юрьевна
Стаж 30
лет
Кандидат медицинских наук
Клиника Доктор АС на Рубцовской набережной
г. Москва, Рубцовская наб., д. 2, корп. 3
Бауманская
1.2 км
8 (499) 519-39-95
8.9
Невролог
Вертебролог
Рефлексотерапевт
Врач высшей категории
Сбитнева Наталья Георгиевна
Стаж 40
лет
Евромедклиник
г. Москва, Сиреневый бульвар, д. 32а
Щелковская
900 м
Первомайская
980 м
8 (495) 185-01-01
8.9
Невролог
Бондарева Анастасия Николаевна
Стаж 10
лет
Медцентр Столица на Бабушкинской
г. Москва, ул. Летчика Бабушкина, д. 48Б
Бабушкинская
1.5 км









8 (499) 519-36-01
Что такое синдром Ли?
Когда врачи диагностируют синдром Ли у ребенка, что это значит, чем характеризуется заболевание, они представляют с трудом. Данным термином называют наследственное нейродегенеративное заболевание, поражающее центральную нервную систему ребенка. Патология характеризуется ранним началом и быстрым прогрессированием, появлением большого количества неврологических симптомов.
Развитие заболевания происходит в результате дефекта множества генов. Локализоваться они могут на Х-хромосоме, аутосомах или ДНК в митохондриях. В зависимости от того, какие изменения имели место, наследование может быть:
- аутосомно-рецессивным;
- сцепленным с полом;
- митохондриальным.
Синдром Ли – классификация
Болезнь Лея, при которой наблюдается развитие рассматриваемого синдрома, может сопровождаться поражением различных компонентов дыхательной цепи митохондрий. В зависимости от этого выделяют:
- Синдром Ли с поражением комплекса НАДН-KoQ-редуктаза. Наследуется рецессивно, митохондриально. В большинстве случаев заболевание обусловлено мутацией генов, расположенных на 19-й, 5-й и 11-й хромосомах. При этой форме болезни наблюдается нарушение процесса переноса электронов и атомов водорода, что ведет к снижению синтеза АТФ.
- Синдром, вызванный дефектами белков в митохондриальном комплексе 2 (сукцинат-KoQ-редуктаза). Данного вида болезнь Лея, генетика патологии связана с мутацией гена на 5-й хромосоме.
- Синдром, развивающийся в результате нарушения структуры белков митохондриального комплекса 3 (KoQН2-цитохром с-редуктаза). Это самый распространенный вариант синдрома Ли, обусловленный мутацией гена на 2-й хромосоме. Характерно аутосомно-рецессивное наследование.
- Синдром, вызванный повреждением митохондриального комплекса 4 (цитохром с-оксидаза). Связан с мутацией генов на 17-й хромосоме или повреждением митохондриальной ДНК.
- Синдром, возникший в результате нарушения структуры комплекса 5 (АТФ-синтаза). В результате наблюдается резкое снижение образования АТФ окислительным путем.
Синдром Ли – причины
Когда у ребенка обнаруживают синдром Ли, генетика в таком случае всегда нарушена. Характер изменений может варьироваться и определяет клинику проявлений болезни. Причинами развития патологии могут выступать мутации большого количества генов, которые расположены на разных хромосомах. Однако сам патогенез этого заболевания в большинстве случаев схож и связан с нарушением процесса клеточного дыхания, нормального функционирования дыхательной цепи митохондрий.
Нарушение структуры белков дыхательной цепи митохондрий ведет к недостаточному синтезу АТФ, которая служит главным источником энергии для всех клеток организма. Большую чувствительность к дефициту энергии испытывают нейроны и нейроглии – элементы нервной системы. Это приводит к развитию необратимых изменений в организме – развивается генетический синдром Ли.
Синдром Ли – частота встречаемости
Синдром Ли – это редкая генетическая патология. Частота ее встречаемости не превышает 1-2 случая на десятки тысяч детей. Точно сказать, как часто возникает заболевание, невозможно – статистика относительно данной патологии не ведется. Подобные расчеты не имеют практической ценности из-за малой частоты встречаемости.

Что такое синдром Оменна
В 1965 году американский генетик Гиберт Оменн (G. S. Omenn) описал данное состояние как семейный ретикулоэндотелиоз с эозинофилией. Позже болезнь была названа синдромом Оменна в честь первооткрывателя. В медицине болезнь имеет другие названия: комбинированный иммунодефицит с гиперэозинофилией и Оменн-синдром. Патология обусловлена нарушениями гуморального и клеточного иммунитета, дисплазией тимуса и аплазией лимфоидной ткани. Клиническая картина синдрома Оменна сходна с симптомами при ТПХ (реакция «трансплантат против хозяина») и проявлениями аутоиммунных заболеваний.
Синдром Оменна (Omenn syndrome) — редкая аутосомно-рецессивная форма ТКИД, вызванная мутацией генов RAG1 и RAG2, локализированных в хромосоме 11р13. Заболевание манифестирует сразу после рождения, характеризуется гепатоспленомегалией, чешуйчатым шелушением и эритродермией кожных покровов, хронической диареей. У грудных детей с синдромом Оменна обнаруживается повышенный уровень иммуноглобулина IgE, лейкоцитоз, высокая концентрация эозинофилов в периферической крови. Также отмечается снижение уровня иммуноглобулинов класса G, А, М, увеличение концентрации Т-лимфоцитов и абсолютное отсутствие В-лимфоцитов. Синдром Оменна — смертельно опасная болезнь, которая поддается коррекции с помощью ТКМ.
Если вам необходима консультация специалиста по орфанным заболеваниям, заполнив все поля формы, и наши консультанты будут рады Вам помочь.
Код МКБ 10
МКБ-10. По международной классификации болезней 10 пересмотра синдром Оменна входит в группу комбинированных иммунодефицитов D81 и имеет код D81.1 (ТКИД с низким содержанием T- и B-клеток).
лечение
Янтарная кислота была изучена, и показала эффективность как для синдрома Leigh, и синдрома MELAS . Высоким содержанием жира, низким содержанием углеводов диеты может следовать , если ген на Х — хромосоме участвует в синдроме индивидуума Leigh. Тиамин (витамин В 1 ) может быть предоставлена , если пируват — дегидрогеназы известно или подозревается. Симптомы лактоацидоза лечат путем дополнения диеты с бикарбонатом натрия (соды выпечки) или цитрат натрия , но эти вещества не лечить причину синдрома Leigh. Дихлорацетат также может быть эффективным при лечении синдрома Leigh-ассоциированного лактацидозу; исследования продолжаются по этому веществу. Коэнзим Q10 добавки были замечены для улучшения симптомов в некоторых случаях.
Клинические испытания препарата EPI-743 для синдрома Leigh продолжаются.
В 2016 году Джон Чжан и его команда в New Hope Fertility Center в Нью — Йорке, США, провели шпиндель переноса митохондриальной донорства технику на мать в Мексике , которая была в опасности производства ребенка с болезнью Leigh. Здоровый мальчик родился 6 апреля 2016 года , однако, пока не уверен в том , что техник полностью надежен и безопасен.
Лечение
К сожалению, на данный момент не существует эффективной терапии синдрома Лея. Лечение направлено на устранение симптомов и продление жизни больного на некоторое время.
Для лечения синдрома Лея используют:
- препараты тиамина (витамин B1);
- рибофлавин (B2);
- дихлорацетат натрия;
- токоферол (E);
- бикарбонат натрия;
- коэнзим Q10;
- димефосфон (для купирования кризов).
Эти препараты, а также низкоуглеводная диета с высоким содержанием жиров, могут положительно влиять на нарушенный энергетический обмен в организме, что несколько замедлит течение заболевания.
В комплексе назначаются средства симптоматической терапии. Например, лекарства для поддержания работы почек, если их функция нарушена из-за синдрома Ли.
На данный момент лекарства от заболевания нет. Но существует препарат EPI-743, который находится на стадии испытания.
В 2016 году впервые была проведена операция «рождения от 3-х родителей», она должна послужить мерой профилактики заболевания у младенца. Методика заключается в применении донорских митохондрий из яйцеклетки.
Прогноз заболевания и профилактика
Прогностическая картина при синдроме Оменна неблагоприятная, в течение 1-2 лет после рождения дети погибают из-за прогрессирующих воспалительных процессов, частого возникновения грибковых, вирусных и бактериальных инфекций на фоне тяжелого иммунодефицита. Единственная возможность продлить жизнь ребенку — это трансплантация костного мозга, после которой уровень выживаемости составляет до 85%. Профилактики от синдрома Оменна не существует, поскольку это генетическое заболевание. Специалисты рекомендуют будущим родителям, которые являются носителями дефектного гена RAG, пройти консультацию генетика перед планированием беременности.
Подходы к диагностике и лечению нейродегенеративных патологий
Ранняя диагностика недуга
Основная задача медицины – научиться определять наличие проблемы на латентной стадии. Возможно, при сохранности большего количества нейронов терапия будет иметь успех. Ведь пока почти все патологии считаются неизлечимыми.










Для решения этой задачи ученые предлагают выделять группы риска. Такая стратификация поможет проводить поиск болезни не в огромной популяции, а в небольшой когорте. Например, при болезни Альцгеймера – это e4 вариант аполипопротеина E. При патологии Паркинсона – это контакт с пестицидами.
Второй вариант для раннего поиска – это изучение биомаркеров. Это молекулы, которые позволяют заранее говорить о болезни. При этом их можно обнаружить на здорового человека без симптомов. Биомаркером при болезни Альйгеймера является амилоид. Его большое количество определяют в ликворе еще до появления клиники.
Гипокситерапия
Гипербарическая оксигенация активно использовалась в середине 20 века. Показаний для проведения сеансов в барокамере довольно много. И болезни с деструкцией нейронов не исключение.
Предположительно лечение кислородом стабилизирует свободные радикалы и уменьшает повреждение клеток. Результаты исследований показали, что метод работает. Но эффективность довольно низкая. Кроме того, имеются ограничения при его использовании.
Гипокситерапия основана на вдыхании газовой смеси с пониженным содержанием кислорода. При проведении сеанса пациент поочередно дышит смесью кислорода и обычного воздуха. Метод доступен во многих санаториях, отделениях реабилитации. Масштабных исследований, позволяющих судить о его эффективности, не проводилось.
Моделирование нейродегенеративных болезней
Тестирование лекарств проводится на животных моделях. Обычно используются лабораторные мыши. Ученые способны выводить поколения грызунов с дефектными генами (с патологией Паркинсона, с накоплением бета-амилоидов в клетках). На трансгенных животных тестируют возможности лекарственной терапии. Однако, часто хороший результат в лаборатории не приводит к успеху в клинике.
Некоторые эксперименты проводятся на клеточных культурах. Сейчас наука может длительно поддерживать жизнеспособность клеток. При этом культуры имеют двух- или трехмерное строение. При этом искусственные нейроны приходится длительно выращивать. А погибают они через 2-3 недели. При моделировании на животном возможно отследить весь процесс болезни от первых симптомов до гибели животного.
Синдром Ли – лечение
Подострая некротизирующая энцефаломиелопатия, связанная с синдромом Ли, практически не поддается лечению. Прежде чем выставить подобный диагноз, пациенту назначают множество обследований:
- МРТ головного мозга;
- Электронейромиографию;
- молекулярно-генетические анализы.
Лечение в большинстве случаев имеет симптоматический характер. Врачи прилагают все усилия, чтобы облегчить состояние пациента. Полное соблюдение всех рекомендаций и назначений медиков гарантирует предупреждение возможных приступов, судорог и других проявлений синдрома. Мероприятия направлены на борьбу с основными симптомами синдрома.
Можно ли вылечить синдром Ли?
Синдром Ли у ребенка не поддается лечению. Специфической терапии подобной патологии не существует. Основу проводимых лечебных мероприятий составляет исключение симптомов болезни, ее проявлений и облегчение самочувствия пациента. С этой целью используются ноотропные, противосудорожные средства.
Важной составляющей частью лечения является применение витаминов, которые служат кофакторами ферментов, входящих в дыхательную цепь митохондрий:
- витамина В1;
- витамина В6;
- витамина Q10.
Признаки болезни у детей
Патология чаще всего начинает проявляться уже в младенчестве. У синдрома Лея клиническая характеристика бывает различной, в зависимости от возрастной группы, к которой относится больной. В целом симптомы заболевания всегда ярко выражены, и это позволяет быстро выявить патологию и начать своевременное лечение. Синдром характеризуется тяжелым течением, причем признаки у взрослых и детей обычно различны.
В возрасте до трех лет у ребенка возникают первые симптомы:
- Тошнота и рвота.
- Задержка двигательного и физического развития.
- Нарушение глотания.
- Быстрая потеря веса.
- Отсутствие аппетита.
- Дрожание конечностей.
- Нарушения зрения.
- Частые ОРВИ.
- Судороги и припадки.
- Учащенное дыхание.
- Нарушения тонуса мышц.
- Головные боли и пониженное давление.
- Нарушения сознания.
Бывают случаи, когда синдром может быть спровоцирован прививкой или ранее перенесенным вирусным заболеванием.

У синдрома Лея тип наследования материнский. Это значит, что все дети, рожденные от женщины, которая является носительницей патологии, будут больны.
Синдром Лея: развитие. Группы риска
Эта патология считается одним из самых опасных заболеваний головного мозга. Чаще всего она возникает у младенцев, реже – у подростков и взрослых. Болезнь может протекать как в острой, так и в хронической форме. Наиболее тяжелым считается подострое течение. К этой патологии необходимо подходить крайне внимательно. Она может провоцировать повреждения головного мозга, ЦНС и стать причиной смерти больного. Наиболее подвержены этому заболеванию новорожденные, грудные младенцы и дети до трех лет.
Синдром имеет генетическую природу. Поэтому к группе риска относятся люди, у которых в роду были случаи подобного заболевания. Если женщина с наследственной предрасположенностью к подобной патологии наблюдается у врача по поводу беременности, она должна обязательно рассказать об этом заболевании. Тогда специалист проведет необходимый осмотр и диагностику и будет принимать решение по поводу дальнейших действий.
Что такое синдром Лея?

Синдром Лея – это наследственное нарушение метаболизма, при котором страдает ЦНС
Патологический процесс, который приводит к развитию синдрома Лея (болезни Ли), связан с нарушением выработки ферментов, ответственных за обеспечение тела человека необходимым количеством энергии.
Заболевание указано в международной классификации болезней МКБ 10 под кодом G31.
Болезнь передается по наследству и приводит к развитию энцефаломиелопатии — поражению головного и спинного мозга.
Причины нарушения
Редкое заболевание развивается из-за нехватки ферментов, обеспечивающих обмен пировиноградной кислоты. Также возникают дефекты в процессах миграции электронов в респираторные цепи. В результате становится недостаточным синтез АТФ, нарушается клеточное дыхание и энергетический обмен.









Возникновению патологии способствуют следующие отрицательные факторы:
- Мутация генов на фоне нарушения работы митохондрий.
- Генетическая обусловленность.
- Доброкачественные или злокачественные новообразования в мозжечке.
- Пролиферативные и воспалительные процессы в сосудах.
- Осложнения дегенеративных заболеваний центральной нервной системы.
- Острые инфекции или вакцинации против них.
Любой из этих факторов может послужить толчком для развития синдрома Лея.
Симптоматика

Синдром Лея вызывает общую слабость, тошноту и рвоту
Заподозрить развитие у человека синдрома Лея можно по ряду характерных симптомов, присутствующих во всех формах заболевания. Клиническая характеристика болезни Ли:
- тошнота в сопровождении рвоты;
- быстрое снижение веса без видимой на то причины;
- замедленное психомоторное развитие;
- снижение интереса к еде;
- дрожь в верхних и нижних конечностях;
- затруднение глотания;
- повышенная утомляемость;
- нарушение координации движений;
- сонливость и апатия;
- снижение остроты зрения; искажение цветовосприятия; слепота вследствие атрофии зрительных нервов;
- затрудненное или аномальное дыхание (дыхание Чейн-Стокса);
- судороги;
- мышечная гипотония;
- головные боли;
- нарушения сознания.
Клинические признаки синдрома возникают обычно на первом году жизни ребенка. Сначала преобладают симптомы неспецифические (отставание в развитии, снижение аппетита и веса и др.). Затем развиваются неврологические нарушения. Сниженный тонус мышц сменяется их спастическим напряжением, повышением сухожильных рефлексов. Может присоединяться миокардиодистрофия. Страдает функция печени.
Осложнения и последствия
Если пациенту с синдромом Лея не будет оказана своевременная медицинская помощь, то болезнь продолжит активно прогрессировать. В результате этого повышается вероятность развития паралича дыхательной системы и инфекционных патологий.
Игнорирование проблемы приводит к летальному исходу.
Методы лечения синдрома Лея
Не всегда можно излечить заболевание, если оно обретает хроническую форму, человек заканчивает летальным исходом. Если вовремя диагностировать синдром, пройти необходимый курс терапии, невропатолог назначит эффективное лечение. При диагностике применяют такие способы:
1. Электроэнцефалография.
2. Обязательно нужно сдать общий анализ крови.
3
Немаловажное значение играет компьютерная томография
4. Дополнительно обследовать головной мозг, используя магнитно-резонансную терапию.
Биохимическое исследование крови показывает, что накапливается пировиноградная и молочная кислоты в ликворе, крови. Увеличивается аланин в крови, повышаются кетоновые тела. В моче повышается экскреция органической, фумаровой кислоты. Также в крови и тканях снижен уровень карнитина.
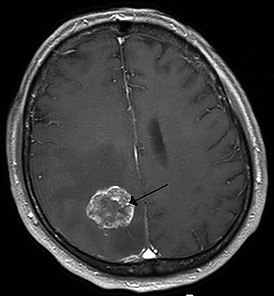
ЭЭГ показывает эпилептическую активность. На МРТ можно увидеть, что расширенные мозговые желудочки, с двух сторон поражен мозг, в базальных ганглиях наблюдается кальцификация. Иногда выявляют атрофию мозга.
При лечении назначают медикаментозные препараты, в составе которых витамин В1. Также советуют принимать антибиотики, лучшим является Биотин, Ампициллин
Важно придерживаться диеты, белка можно съедать не больше, чем 1 грамм
Синдром Лея является опасным заболевание, которое возникает в разном возрасте, чтобы защититься от него, необходимо ежедневно выполнять специальный комплекс гимнастики, так можно улучшить состояние центральной нервной системы
Важно тщательно заботиться о своем организме при синдроме в профилактических целях
Симптомы и признаки
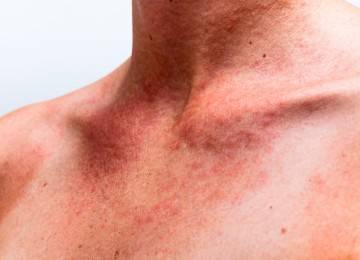
У малышей с синдромом Оменна симптоматическая картина проявляется рано, в течение первого года жизни, часто сразу после рождения. Расстройства иммунитета сопровождаются постоянными пневмониями, хронической диареей, грибковыми поражениями кожи и слизистых оболочек. Нередко возникает покраснение, сыпь и шелушение кожного покрова. Также синдром Оменна сопровождается снижением аппетита, что приводит к плохой прибавке массы тела. У детей наблюдается частое повышение температуры (лихорадка), отиты, гиперплазия лимфоузлов, одновременное увеличение селезенки и печени.









На фоне экссудативной эритродермии и хронической диареи снижается содержание общего белка в крови, что приводит к нарушениям обмена веществ и генерализованным отекам. Воспалительные симптомы нередко вызывают алопецию, выпадение бровей и ресниц. Также при синдроме Оменна может развиваться колобома глаз (аномалия в глазной оболочке), синдром CHARGE, митохондриальная миопатия, гипоплазия волос и хрящей.
Признаки и симптомы
Симптомы синдрома Leigh классически описывается как начало в младенчестве и приводит к смерти в течение промежутка нескольких лет; Однако, как все больше случаев признаются, то очевидно, что симптомы могут возникать в любом возрасте, в том числе в подростковом возрасте или в зрелом возрасте-и пациенты могут выжить в течение многих лет после установления диагноза. Симптомы часто первым видели после инициирующего события, что налоги производства энергии в организме, такие как инфекция или хирургическое вмешательство. Общий курс синдрома Leigh является одной из эпизодических регрессии развития в периоды метаболического стресса. Некоторые пациенты имеют длительные периоды без прогрессирования заболевания в то время как другие развиваются прогрессирующее снижение.
У детей с синдромом имеют симптомы , которые включают в себя понос , рвота и дисфагия (проблемы с глотанием или сосание), что приводит к неспособности процветать . Дети с ранней стадией заболевания Leigh также могут появиться раздражительность и плакать гораздо больше , чем обычно. Приступы часто видели. Избыток лактата может быть замечен в моче , цереброспинальной жидкости и крови человека с синдромом Ли.
По мере прогрессирования заболевания, то мышечная система ослаблена по всему телу, так как мозг не может контролировать сокращение мышц. Гипотония (низкий мышечный тонус и сила), дистония (непроизвольное, устойчивое сокращение мышц) и атаксия (отсутствие контроля за движением) часто встречаются у людей с болезнью Leigh. В глаза особенно чувствительны; мышцы , которые контролируют глаза становятся слабыми, парализованы или неконтролируемое в условиях , называемых Офтальмоплегия (слабость или паралич) и нистагм (непроизвольные движения глаз). Медленные саккады также иногда видели. Сердце и легкие также может потерпеть неудачу в результате болезни Leigh. Гипертрофическая кардиомиопатия (утолщение части сердечной мышцы) также иногда встречается и может привести к смерти; также ассоциируется с синдромом Ли. У детей с синдромом Ли-ассоциированные перегородочных дефектами желудочковых , вызванными пирувата — дегидрогеназой, высоким лбом и большими ушами видно; лицевые аномалии не типичны для синдрома Leigh.
Однако, дыхательная недостаточность является наиболее частой причиной смерти у людей с синдромом Ли. Другие неврологические симптомы включают периферическая нейропатия , потеря чувствительности в конечностях , вызванных повреждением периферической нервной системы .
Гипертрихоз рассматривается при синдроме Ли , вызванной мутацией в гене ядерного SURF1 .
Причины
Катализатором развития синдрома Оменна являются мутации генов RAG1 и RAG2, участвующих в перестройке антигенных рецепторов в- и т-лимфоцитов. RAG-дефекты приводят к полной блокировке развития Т- и В-клеток, что вызывает тяжелую комбинированную иммунную недостаточность с отсутствием зрелых форм в- и т-лимфоцитов.
Синдром Оменна наследуется по аутосомно-рецессивному типу, то есть дети могут родиться с заболеванием, если оба родителя являются носителями мутантного гена. Риск рождения ребенка с данным синдромом составляет 25 %. Частота наследования генного дефекта у мальчиков и девочек одинаковая. Симптоматическая картина и развитие реакции ТПХ обусловлены разрушением тканей и органов на фоне поступления в кровь пролиферирующих материнских лимфоцитов во внутриутробный период. При большом количестве материнских лимфоцитов в организме плода развиваются множественные некрозы в селезенке и печени.
патофизиология
Характерные симптомы синдрома Leigh, по крайней мере , частично вызвана двусторонними, очаговых поражений в стволе головного мозга , базальных ганглиев , мозжечка и других областей мозга. Очаги принимать различные формы, в том числе областей демиелинизации , спонгиозы , глиозом , некроза и капиллярной пролиферации. Демиелинизация является потерей миелиновой оболочки вокруг аксонов нейронов, подавляя их способность общаться с другими нейронами. Ствола мозга участвует в поддержании основных жизненных функций , таких как дыхание, глотание и обращения; базальные ганглии и мозжечок движение контроля и баланс. Повреждение этих областей , следовательно , приводит в основных симптомах синдрома Leigh-потере контроля над функциями , контролируемых этими областями.
Лактоацидоз иногда ассоциируется с синдромом Ли обусловлен накоплением пирувато , который не может быть обработано у людей с определенными типами окислительных фосфорилирования недостатков. Пируват либо преобразуется в аланин с помощью аланинаминотрансферазы или преобразованы в молочную кислоту с помощью лактата deydrogenase ; оба этих веществ может затем накапливаться в организме.
Диагностика

Для подтверждения диагноза назначают МРТ головного мозга
При подозрении на данное заболевание требуется проведение следующих методов диагностики:
- Магнитно-резонансная томография (МРТ). Позволяет выявить поражение отделов мозга.
- Электронейромиография (дает возможность оценить степень разрушения миелиновой оболочки нервов).
- Молекулярно-генетическая диагностика.
- Электрокардиография. Предназначена для изучения состояния сердечно-сосудистой системы.
- Офтальмоскопия глазного дна. Позволяет выявить причину нарушения зрения и наличие аномальных изменений в органе.
- Может потребоваться забор ликвора. Эта процедура помогает определить уровень белка и наличие лимфоцитарного плеоцитоза.
- Биохимический анализ крови позволяет определить повышенное количество пировиноградной и молочной кислот (лактат-ацидоз), кетоновых тел.
При постановке диагноза требуется исключить развитие у пациента заболеваний со схожей симптоматикой: лейкодистрофии, энцефаломиелита инфекционно-аллергического генеза, амавротической идиотии.