Сопутствующие заболевания
Сопутствующие нарушения в том же спектре заболеваний , как HDLs включают болезнь Наса-Hakola ( поликистозный lipomembranous osteodysplasia с склерозирующей лейкоэнцефалопатией ), а также тип лейкодистрофия с пигментными заполненными макрофагами под названием пигментной ортохроматический лейкодистрофия (Põld). В дополнении к болезни белой материи, Нас-Hakola вызывает костные кисты. Это вызвано мутациями в генах , участвующих в одной и той же колониестимулирующий фактор (CSF) сигнального пути каскада , как определено в ЛВП.
Болезнь Нас-Хакол по- видимому, вызвана мутациями в Тайро белке тирозинкиназа-связывающий белке ( TYROBP — также известный как DAP12) или запускающего рецептор экспрессируется на миелоидные клетки (2 TREM2 ) белка. В то время как различные мутации гена происходят в пределах пути для Наса-Hakola и ЛВПА, и характеризуется дегенерацией белого вещества с аксонами сфероидов. Современные исследователи в этой области считают , что более глубоким анализом и сравнением двух генетических аномалий в этих нарушениях может привести к лучшему пониманию механизмов болезни в этих редких заболеваниях. PÕLD демонстрирует невоспалительные демиелинизации аксонов с начальными симптомами эйфории, апатия, головная боль и исполнительной дисфункции . В то время как ЛВП является аутосомно — доминантным, некоторые семьи с PÕLD имеют особенности , которые предполагают аутосомно рецессивное наследование. Тем не менее, PÕLD недавно было показано, что один и тот же генетический базис как ЛВП.
диагностика
Исследования в 2012 включает в себя исследование микроглии функции. Эта работа будет способствовать дальнейшему выяснить, является ли эта болезнь в основном дефект функции микроглии. Для такого исследования, клетки микроглии из HDLs родственных можно культивировать из мозга аутопсии и анализировали в сравнении с нормальными клетками микроглии на основе различий в мутационных событий и экспрессии фактора роста.
Клинические и генеалогические исследования
Для того, чтобы получить лучшее понимание болезни, исследователи ретроспективно проанализировали медицинские записи пробандов и другим , которые были оценены с помощью клинических исследований или вопросников. Образцы крови собирают из семей пробандов для генетического тестирования. Эти члены семьи оценивают по их стандартную медицинскую историю , на их прогрессирование как симптомов болезни Паркинсона ( Унифицированная оценки болезни Паркинсона Scale ), и на их прогрессирования когнитивных нарушений , таких как деменция ( Folstein Test ).
Neuroimaging
Стандартный МРТ сканирование было выполнено на 1,5 Тесла сканеров с толщиной 5 мм и шагом 5 мм для скрининга белого вещества поражений в определенных семьях. Если интенсивность сигнала в МРТ в белых районах вещества выше , чем в серых областях материи, пациент считаются риском ЛВПА, хотя ряд других заболеваний , также может производить белые изменения материи и выводы не являются диагностическими без генетического тестирования или патологическое подтверждение.
патология
Тканевые срезы биопсии головного мозга или головного мозга аутопсии , как правило , встроен в парафин , из которого секции вырезать смонтированы на предметные стекла для гистологических исследований. Окрашивание для миелина и аксонов патологии показывают патологические изменения, характерные для ЛВП определены в белом веществе неокортекса , базальных ганглиев , таламуса , среднего мозга , мосте и спинного мозга. В дополнение к обычным гистологических методов ( Н & Е окрашивания ), образцы оценивали с иммуногистохимии для убиквитина , амилоидного белка — предшественника, и нейрофиламентов , чтобы охарактеризовать аксонов изменения и основной белок миелина для миелина патологии. Иммуногистохимических пятна для микроглии (CD68 или HLA-DR) и астроцитов (GFAP), также полезные методы для характеристики белого вещества патологии. С аналогичной патологией в PÕLD, ЛВП обычно группируются во взрослом возрасте лейкоэнцефалопатии с аксонами эллипсоидов и пигментированной глией (ALSP), чтобы дать этим отдельно в соответствии с признанными условиями повышенного внимания.
классификация
ЛВП подпадает под категорию мозга заболеваний белого вещества под названием leukoencephalopathies, которые характеризуются определенной степенью дисфункции белого вещества. ЛВП имеет повреждения белого вещества с нарушениями в миелиновой оболочки вокруг аксонов, где причинные влияния в настоящее время постоянно проводятся исследования на основе последних генетических исследований. Исследования Sundal и коллегами из Швеции показали , что аллель риски кавказцев может быть причинной , поскольку случаи , выявленные до сих пор были среди крупных кавказских семей.
Диагностика
 Диагностика лейкодистрофии
Диагностика лейкодистрофии
Для определения разновидности лейкодистрофии головного мозга потребуется комплексный подход, который основывается на инструментально-лабораторных исследованиях.
Однако не последнее место занимает первичная диагностика, которая включает в себя:
- изучение истории болезни как маленького пациента, так и его родителей – для выяснения пути наследования патологии;
- тщательный физикальный осмотр – для оценивания тонуса мышц, рефлексов, походки и координации движений. Сюда также стоит отнести консультации ЛОР-врача и офтальмолога – для определения наличия нарушений со стороны зрения или слуха;
- детальный опрос родителей пациента – для выяснения первого времени появления специфических признаков, поскольку в некоторых случаях очень важная информация касательно того, возникла ли симптоматика в младенческом возрасте или в ювенильном периоде.
Лабораторные исследования ограничиваются:
- анализом спинномозговой жидкости;
- биохимическими анализами крови – для выявления того, какие патологические вещества скапливаются при протекании того или иного варианта недуга.
Инструментальная диагностика уточняет тип болезни при помощи таких процедур:
- нейросонография;
- эхо-энцефалография;
- КТ и МРТ.
Также разработаны специфические методики ДНК-диагностики, обнаруживающие подобное заболевание ещё на этапе внутриутробного развития плода. В таких случаях необходима консультация специалиста по генетике.
Миша Фролов, 13 лет, редкое генетическое заболевание – адренолейкодистрофия головного мозга (Апрель 2020).

Адренолеодистрофия: редкое генетическое (наследственное) расстройство, характеризующееся расстройством или потерей миелиновой оболочки, окружающей нервные клетки в головном мозге и прогрессирующей дисфункцией надпочечников. Adrenoleukodystrophy (ALD) является одной из групп генетических заболеваний, называемых лейкодистрофиями, которые вызывают повреждение миелиновой оболочки нервных волокон в головном мозге. Миелиновая оболочка — жирное покрытие, которое действует как электрический изолятор.
Существует несколько форм ALD:
- Классическая форма детства, которая является самой серьезной и затрагивает только мальчиков, может возникать в возрасте от 4 до 10 лет. Она затрагивает только мальчиков, потому что ген находится на Х-хромосоме. Особенности этой формы могут включать в себя визуальную потерю, нарушения зрения, судороги, дизартрию (слабо выраженная речь), дисфагия (затруднение глотания), глухота, нарушения походки и координации, усталость, прерывистая рвота, меланодермия (повышенная пигментация кожи) и прогрессирующая деменция, Наиболее распространенными симптомами обычно являются поведенческие изменения, такие как аномальный уход или агрессия, плохая память и плохая успеваемость в школе.
- Женщины-носители: Еще одна форма АЛД иногда наблюдается у женщин, которые являются носителями расстройства. Симптомы мягкие и могут включать спастический парапарез нижних конечностей, атаксию, гипертонию (чрезмерный мышечный тонус), мягкую периферическую невропатию и проблемы с мочеиспусканием.
- Более мягкая форма для взрослых начинается обычно в возрасте от 21 до 35 лет. Симптомы могут включать в себя ночную жесткость, прогрессирующий спастический парапарез (жесткость, слабость и / или паралич) нижних конечностей и атаксию. Хотя взрослый ALD прогрессирует медленнее, чем классическая форма детства, это также может привести к ухудшению функции мозга.
- Новорожденный (новорожденный) ALD поражает как мужчин, так и женщин. Симптомы могут включать умственную отсталость, лицевые аномалии, судороги, дегенерацию сетчатки, гипотонию (низкий мышечный тонус), гепатомегалию (увеличенную печень) и дисфункцию надпочечников. Эта форма обычно быстро прогрессирует.
Лечение всех форм АЛД является симптоматическим и поддерживающим. Физическая терапия, психологическая поддержка и специальное образование могут быть полезны для некоторых людей.

Прогноз для пациентов с АЛД обычно плохой из-за прогрессирующего неврологического ухудшения. Смерть обычно происходит через 1-10 лет после появления симптомов.
Классификация
В зависимости от экспрессивности (степени проявления) пораженного гена, возраста пациента, в котором произошел дебют болезни, выделяют несколько форм адренолейкодистрофии:
- церебральная (детская, юношеская, взрослая формы) – чаще встречается у детей и подростков, начинается в раннем школьном возрасте, быстро прогрессирует, в большинстве случаев к церебральной форме присоединяется надпочечниковая недостаточность;
- адреномиелонейропатия – характерна для взрослых, начинается в возрасте 12-50 лет, сочетает поражение надпочечников и периферическую нейропатию;
- надпочечниковая недостаточность – встречается в 10% случаев, характерна для мальчиков, снижается продукция глюкокортикоидов, позже – минералокортикоидов;
- неврологическая симптоматика у гетерозиготных носительниц – встречается у женщин, имеющих одну пораженную Х-хромосому и одну здоровую, преимущественно протекает в церебральной форме или в виде адреномиелонейропатии.
Этиология
Лейкодистрофия, или прогрессирующий склероз мозга, получила своё название оттого, что в патологический процесс вовлекается белое вещество этого органа. На сегодняшний день известно большое количество форм болезни, отличающихся видом генетической мутации и возрастной категорией, в которой происходит манифестация симптомов.
Наиболее частые типы болезни, например, метахроматическая лейкодистрофия, диагностируются у одного младенца на сто тысяч новорождённых. Однако есть такие виды патологии, которых зарегистрировано не больше нескольких сотен.
Основной причиной любого вида недуга является генетическая аномалия того или иного фермента. Разновидности и локализация мутировавших генов установлены только для наиболее частых форм патологии.
Зачастую лейкодистрофия характеризуется аутосомно-рецессивным путём наследования, но некоторые виды могут передаваться сугубо по половой принадлежности, т. е. от матери к дочери или от отца к сыну.
Генетически обусловленный дефект наиболее часто приводит к нарушению обменных процессов, что чревато скоплением того или иного вещества в организме. В основном поражаются следующие органы:
- головной мозг;
- почки;
- спинной мозг;
- печень.
Следствием обменного нарушения является:
- разрушение миелина в оболочках нервных стволов;
- гибель или атрофия нейронов;
- замещение погибших нейронов глиальной тканью, которая постоянно разрастается.
По морфологическим признакам лейкодистрофия характеризуется:
- диффузным или симметричным расположением областей гибели миелина в обоих полушариях головного мозга;
- скоплением большого количества продуктов, выделяющихся после распада миелина;
- усиленным разрастанием глии.
Для всех групп болезни характерно развитие в раннем детском возрасте, ещё до того, как ребёнок пойдёт в школу.
Эффективно ли масло лоренцо в качестве альтернативного лечения адренолейкодистрофии?
Ряд недавних исследований показал, что смесь олеиновой кислоты и эруковой кислоты, известная как масло Лоренцо, которая предоставляется мальчикам с БАС до появления симптомов, может предотвратить или отсрочить прогрессирование заболевания. Пока неизвестно, можно ли использовать масло лоренцо для лечения адреномиелоневропатии (версия ALD для взрослых). Кроме того, было доказано, что масло Лоренцо не оказывает никакого влияния на мальчиков с ALD, у которых есть симптомы.
Трансплантация костного мозга оказалась успешной у лиц с адренолейкодистрофией ранней стадии (ALD). Тем не менее, эта процедура несет в себе риск смерти и не рекомендуется для тех, у кого острая или тяжелая стадия ALD, для тех, кто испытывает симптомы в зрелом возрасте, или в неонатальных случаях.
- Определить здоровье организма с помощью кала
- От солнечной аллергии к водной аллергии, почему?
- Что случилось во время нашей смерти?
Адренолейкодистрофия (ALD), инвалидизирующее редкое заболевание
Rated 5/5
based on 2235 reviews

Диагностика
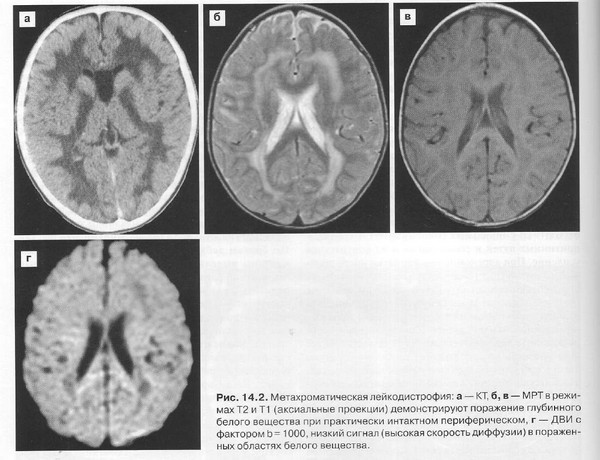
Обычно раздел диагностики принято помещать после клинической картины и перед лечением. Но липидозы, в связи с полиморфизмом симптомов и неуклонным прогрессированием до сих пор, несмотря на существование визуализирующих методов, таких, как КТ и МРТ, часто не поддаются прижизненной диагностике. Нужно помнить, что никакое исследование, даже МРТ с контрастированием в динамике, не в состоянии подтвердить этот процесс, а только «не исключить».
Это связано с тем, что симптомы лейкодистрофий чрезвычайно разнообразны, особенно у ребенка. Логика подсказывает, что скорее возможны с наибольшей степенью вероятности многие другие заболевания. У ребенка это могут быть проявления перинатального поражения центральной нервной системы, детского церебрального паралича, нейроинфекции. Как у детей, так и у взрослых, прежде всего врачам нужно исключать системные инфекционно – аллергические и аллергические поражения, опухоли.
Настоящие трудности возникают при попытке дифференцировать лейкодистрофии от (оптикомиелита Девика, острого рассеянного энцефаломиелита, рассеянного склероза). На помощь могут прийти количественные анализы на определение дефектных липидов в крови и спинномозговой жидкости, а также малодоступные и очень дорогие генетические прицельные исследования, поскольку картина на МРТ не дает однозначного ответа.
Так, врач невролог, видя, например, фото кровоизлияния мозга, сразу поставит диагноз по характерным признакам
При наличии демиелинизирующих очагов нужно обращать внимание на клиническую картину, но, учитывая быстроту прогрессирования дистрофий белого вещества, и отсутствие разработанного лечения, зачастую оказывается, что окончательный диагноз выставляется только на вскрытии, особенно у маленьких детей. .
Клиническая картина
Симптомы поражения очень многообразны. У ребенка можно встретить:
- диффузное снижение мышечного тонуса, с последующей сменой на гипертонус;
- появление дрожания головы, конечностей;
- судороги, немотивированное возбуждение, постоянный крик;
- глазодвигательные нарушения: косоглазие, нистагм, офтальмоплегия, как внутренняя, так и наружная;
- обратное развитие (дети утрачивают все приобретенные навыки);
На поздней стадии развиваются тяжелые параличи конечностей, . Смерть наступает от паралича дыхательной мускулатуры, сосудодвигательного и дыхательного центра продолговатого мозга.
Метахроматическая лейкодистрофия может развиться в юношеском возрасте и у взрослых. В этом случае будут беспокоить:
- мозжечковые и (тремор, гиперкинезы, ригидность);
- атрофия зрительных нервов;
- центральные параличи и парезы;
- возникает выраженная деменция, возникают такие симптомы, как , или расстройство речи.
Нужно помнить, что метахроматическая лейкодистрофия иногда оставляет своим жертвам наибольшее время для жизни, но это время человек проживает тяжелым инвалидом, часто лишенный возможности не только передвигаться, но и мыслить.
О терапии
Специфического лечения даже такого длительно текущего заболевания, как метахроматическая лейкодистрофия во взрослой форме, не говоря уже о быстрых вариантах, поражающих структуры мозга ребенка, не существует. Существующее лечение сводится к введению гормонов, витаминов, поддержания функций мозга, пока человек может дышать.
Единственный шанс восстановить миелин и улучшить работу мозга в наше время – это аутотрансплантация стволовых клеток. Но даже при этом нужно длительный срок для его синтеза (по данным МРТ, год или два). Чаще всего, срок жизни при болезни значительно короче, особенно у детей.
Сравнить оголенные нервы, лишенные миелиновых оболочек, упакованные плотными пучками, можно лишь с энергосистемой целого города, провода и кабели которого не имеют изоляции и скручены между собой. В результате возникнет вспышка короткого замыкания с разрушением всей энергоструктуры. То же самое происходит при этих заболеваниях.

Что такое липидоз?
Нервная система человека является высшим органом, координирующим вегетативные функции (питание, кровообращение, выделение, дыхание), сознательные мышечные движения, обеспечивающие взаимодействие человека с внешней средой. Венцом является высшая нервная деятельность и человеческое мышление, благодаря которому у вас появилась возможность читать этот текст на экране компьютера. Все это невозможно без нервного импульса, который создается нейронами, составляющими серое вещество коры больших полушарий головного мозга, подкорковых ядер и спинного мозга.
Нервные импульсы, которые ежесекундно миллионами генерируют наши нейроны, должны проводиться четко и без потери информации. Это значит, что белое вещество головного мозга, или аксоны – проводники, должны иметь очень хороший «изолирующий слой». Таким изолятором служит липидное вещество миелин, из которого состоят наружные оболочки нервов. Проводящий ток осевой цилиндр нерва плотно обернут несколько раз .
Именно потому, что липиды нерастворимы в воде, их мембраны полностью исключают потери импульсов, которые генерируются в водной среде цитоплазмы нейрона. Известно, что волна возбуждения, которая генерируется в нейроне с помощью работы натриево-калиевого насоса, может распространяться со скоростью более 100 метров в секунду.
Поэтому в нервной системе человека сосредоточено много миелина, который относится к липидам. Нарушение обмена и структуры липидов в головном мозге составляют заболевания, которые называют липидозами. Сюда входит группа наследственных лейкодистрофий веществаголовного мозга, о которых и пойдет речь.
Группа наследственных лейкодистрофий — это очень редкие заболевания, поэтому у врачей есть множество причин вначале подумать о более часто встречающейся патологии.
О наследственных лейкодистрофиях
В неврологии до середины 80-х годов XX века был принят термин «прогрессирующий склероз», в наше время он заменен более точным термином «лейкодистрофия». Лейкодистрофия – это группа заболеваний наследственного характера, которые характеризуются прогрессирующим поражением белого вещества, как головного, так и спинного мозга с разрастанием глиальных элементов и нарушением проведения нервного импульса.
Клиническая картина заболевания
Данное заболевание характеризуется наличием определенной симптоматикой. Симптомы адренолейкодистрофии достаточно ясно говорят о наличии нарушений, происходящих в головном мозге и железах внутренней секреции:
- эйфория;
- депрессия;
- нарушения памяти;
- нарушения походки, движений;
- судорожные приступы;
- расстройства зрения;
- резкие перемены настроения.
Лабильность нервной системы особенно характерна для данного заболевания, когда у больных резко может наступать переход от расслабленного, эйфоричного приятного настроения к нарастанию угнетенного, мрачного состояния, появлению депрессий. Нарушения движений и походки сопровождается, в свою очередь, также явлениями квадриплегии, когда может наблюдаться парез всех конечностей или же отмечается повышенный их тонус, вплоть до появления судорожных приступов.
Со стороны зрительного аппарата, кроме снижения остроты зрения, наблюдаются парезы зрительных мышц, снижение реакций зрачка, атрофия зрительного нерва. Может наблюдаться частичная или полная потеря зрения.
Позже происходит присоединение нарушений со стороны нервной системы, когда, кроме изменений поведения и интеллекта, наблюдаются симптомы поражения ЦНС и психические отклонения. Снижается успеваемость в школе, наступает дизартрия, появляются парезы спастического характера.
Нарастают признаки слабоумия и развивается деменция. Крайняя степень таких нарушений выражается в развитии психического маразма. Обусловлены эти явления процессами распада в структуре головного мозга (таламусе, паллидуме, путамене).
Спастические проявления при данном заболевании прогрессируют, в результате чего могут образовываться параличи конечностей, с полной потерей их чувствительности. Наблюдаются нарушения речи, атаксия. Кроме того, выраженным симптомом адренолейкодистрофии является гипогонадизм, то есть недоразвитие половых желез, которые имеют маленькие размеры и, соответственно, сниженные функции. Отмечается также повышенная пигментация кожи.
лечение
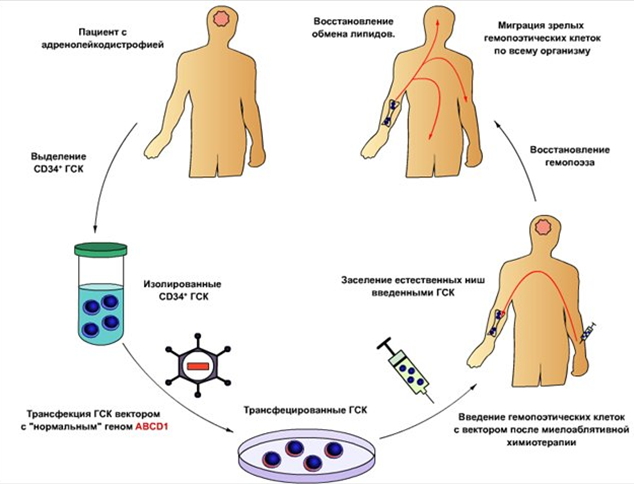
Со многими различными типами лейкодистрофии и причины, методы лечения лечения различны для каждого типа. Многие исследования и клинические испытания в процессе , чтобы найти лечение и терапию для каждого из различной лейкодистрофия. Стволовые клеточные трансплантаты и генная терапия , как представляется, наиболее перспективным при лечении всех Leukodystrophies условии , что оно сделано как можно раньше. Для hypomyelinating лейкодистрофии, терапевтическое исследование в клеточной терапии представляется перспективным. Олигодендроцитов клетки — предшественники и нейронные стволовые клетки были успешно пересажены и показали , чтобы быть здоровым годом позже. Дробная анизотропия и радиальные карты температуропроводности показали возможные миелинизации в области трансплантации. Индуцированные плюрипотентные стволовые клетки , клетки — предшественники олигодендроцитов, коррекция генов, и трансплантации в целях содействия созревания, выживание и миелинизации олигодендроциты , как представляется, первичные маршруты для возможных обработок.
Для трех типов лейкодистрофии ( Х-хромосомой адренолейкодистрофия (Х-ALD), метахроматическая лейкодистрофия (MLD) и Краббе болезнь (глобоидные клетки лейкодистрофия — ООВ), генной терапии с использованием аутологичных гемопоэтических стволовых клеток для переноса гена заболевания с лентивирусов векторов показали быть успешным и в настоящее время используются в клинических испытаниях для X-ALD и УСУ. прогрессирование X-ALD показал нарушаться с гемопоэтических стволовых генов клеточной терапии , но точная причина , почему демиелинизации останавливается и количество стволовых клеток необходимо неясна . в то время как происходит накопление очень длинноцепочечные жирные кислот в головном мозге, это не кажется , чтобы быть причиной заболевания , как генная терапия не приводит его.
Аденосателлитные векторы также были использованы в внутримозговых инъекций для лечения MLD. У некоторых пациентов с УСОЙ, их IQ увеличивается, нервная проводимость улучшилась, их MRIs оказались стабильными, и имел нормальные уровни фермента. Хотя большая часть пациентов , кажется, улучшается после пересадки, некоторые не очень хорошо реагируют на лечение, которое может привести к разрушительным результатам. Для тех лейкодистрофии , которые являются результатом дефицита лизоцима ферментов, таких как Болезнь Краббе , замена энзимотерапии кажется надежд, однако, это доказывает трудным , так как гематоэнцефалический барьер сильно ограничивает то , что может пройти через в центральную нервную систему. В связи с этим препятствием, большинство исследований и клинических испытаний обращаются к аллогенной трансплантации гемопоэтических стволовых клеток.
Лечение
Диетическая терапия
Первоначальные попытки диетической терапии в ALD участвует ограничивая потребление очень-длинноцепочечных жирных кислот (VLCFA). Диетическое потребление не является единственным источником для VLCFA в организме, так как они также синтезируется эндогенно. Это диетическое ограничение не влияет на уровни VLCFA в плазме крови и других тканях организма. После того, как осознание того, что эндогенный синтез является важным вклад в VLCFA в организме, усилия по диетической терапии сдвинуты к ингибированию этих путей синтеза в организме. В родители из Лоренсо Одон , мальчик с ALD, направляла усилия по разработке диетического лечения , чтобы замедлить прогрессирование заболевания. Они разработали смесь ненасыщенных жирных кислот ( глицерин , триолеат и глицерил trierucate в соотношении 4: 1), известные как масло Лоренца , который ингибирует удлинение насыщенных жирных кислот в организме. Supplementation маслом Лоренцо было установлено, нормализации концентрации VLCFA в организме, хотя его эффективность при лечении церебральных проявлений заболевания до сих пор спорными и недоказанными. Опыты с маслом Лоренца показали , что это не останавливает неврологическое ухудшение симптоматических пациентов, а также не улучшает функцию надпочечников.
пересаживать
В то время как диетическое лечение было показано , чтобы быть эффективным , чтобы нормализовать очень-длинные концентрации жирных кислот в плазме пациентов с ALD, является единственным лекарством , которое может остановить демиелинизации , что является отличительной чертой церебральных форм болезни. Для того , чтобы быть эффективными, пересадка должна быть сделана на ранней стадии заболевания; если демиелинизация прогрессировала, трансплантат может ухудшить исход, и увеличить скорость снижения. В то время как трансплантаты , как было показано , чтобы быть эффективными при остановке процесса демиелинизации в тех , которые представляют с детства церебральной формы АБП, последующие из этих пациентов показал , что он не улучшает функцию надпочечников.
Генная терапия
Для пациентов , у которых не может быть найден подходящий матч для пересадки, были исследования в использовании генной терапии . Соответствующие векторы , которые выбраны и модифицированы для экспрессии дикого типа ABCD1 , который затем трансплантировали в пациентах с использованием аналогичной процедуры, что и для костного мозга или трансплантации стволовых клеток. Генная терапия только опробована на небольшом числе пациентов, главным образом в Франции . Эти пациенты были рассмотрены только для генной терапии после того, как не было HLA матча для традиционной трансплантации. В двух зарегистрированных случаях, генная терапия была успешной, с разрешением процесса демиелинизации до двух лет после процедуры. Несмотря на то, генной терапии была успешной в решении неврологических симптомов, плазменные уровни VLCFA оставались повышенными.
недостаточность надпочечников
Лечение надпочечниковой недостаточности, которая может сопровождать любого из распространенных мужских фенотипов ALD не решает ни одной из неврологических симптомов. Гормональный является стандартной для пациентов ALD, демонстрирующих недостаточность надпочечников. Недостаточность надпочечников не решает с успешной трансплантацией; большинство пациентов все еще требуют замены гормона.
Профилактические меры
Планирование беременности и медико-генетическая консультация будущих родителей, имеющих в семье случаи заболевания адренолейкодистрофией, является наиболее эффективной мерой первичной профилактики заболевания. Врач-генетик определяет вероятность рождения ребенка с патологией, учитывая семейный анамнез родителей.
Во время беременности возможна пренатальная диагностика плода биохимическими методами или исследованием ДНК.
При подозрении на заболевание пациенту проводят полную диагностику с целью выявления заболевания на доклинической стадии, когда диетотерапия и медикаментозная коррекция дает хороший результат и возможность продлить полноценную жизнь пациенту.
Источники информации:
1. Федеральные клинические рекомендации по диагностике и лечению Х-сцепленной адренолейкодистрофии. — 2013.
2. Адренолейкодистрофия в сочетании с адреномиелоневропатией и лечение больного пероральным масло Лоренцо/ Евтушенко С.К., Евтушенко И.С.// Международный неврологический журнал. — 2012. — №5.
3. Х-сцепленная адренолейкодистрофия: клинико-биохимический и молекулярно-генетический анализ: Автореферат диссертации/ Ломоносова Е.З.- 2006.
4. Х-сцепленная адренолейкодистрофия: некоторые сведения о заболевании/ Еремина E.P.// Вестник Бурятского государственного университета. – 2015.
Симптомы Х-сцепленной адренолейкодистрофии
Симптомы могут варьироваться в зависимости от типа АЛД.
Х-сцепленная АЛД (детская церебральная АЛД)
Эта форма является наиболее тяжелой. Она встречается только у мальчиков. Симптомы обычно начинаются между 2-10 годами. Около 35% пациентов может иметь тяжелые симптомы на ранней стадии. В среднем, смерть наступает в течение двух лет. Некоторые пациенты могут жить несколько десятилетий.
Первоначальные симптомы включают в себя:
- Поведенческие изменения;
- Ухудшение памяти.
По мере прогрессирования болезни, развиваются более серьезные симптомы. К ним относятся:
- Потеря зрения;
- Приступы;
- Потеря слуха;
- Затруднения при глотании и разговоре;
- Трудности при ходьбе и координации движений;
- Рвота;
- Усталость;
- Усиление пигментации («загара») на коже, из-за дефицита гормона надпочечников (болезнь Аддисона);
- Прогрессивное слабоумие;
- Вегетативное состояние или смерть.
Адреномиелоневропатия (АМН)
Это наиболее распространенная форма. Симптомы АМН могут появиться после 20-ти лет. Болезнь прогрессирует медленно и может включать:
- Слабость, неловкость, потеря веса, тошнота;
- Эмоциональные нарушения и депрессии;
- Проблемы с мышцами (например, проблемы при ходьбе);
- Половые проблемы или импотенция;
- Дисфункция надпочечников;
Х-сцепленная АЛД (церебральная АЛД взрослых)
При этом типе, симптомы обычно редко проявляются в молодом (до 20 лет) или среднем возрасте (до 50 лет). Эта форма АЛД вызывает симптомы, похожие на шизофрению и слабоумие. Обычно болезнь быстро прогрессирует. Смерть или вегетативное состояния может наступить через 3-4 года.
Гетерозиготная форма АЛД
Эта форма встречается только у женщин. Симптомы могут быть умеренными или тяжелыми. На функционирование надпочечников гетерозиготная форма АЛД обычно не влияет.
Симптомы
Характер проявления генной активности определяет разнообразие клинической картины и степень выраженности симптомов.
Для церебральной формы характерны следующие нарушения:
- гиперактивное поведение или снижение поведенческой активности;
- проблемы в обучении, снижение памяти, недостаток концентрации внимания;
- эпизоды агрессии;
- быстро прогрессирующее слабоумие;
- нарушения зрительной функции и слуха;
- позднее присоединяются симптомы надпочечниковой недостаточности;
- смертельный исход наступает в течение 15 лет после начала проявления симптоматики.
- Надпочечниковая недостаточность характеризуется:
- гиперпигментацией кожных покровов;
- хронической усталостью, слабостью мышц;
- гипогликемией;
- тошнотой, головной болью, низким артериальным давлением.
Адреномиелонейропатия характеризуется сочетанием хронической надпочечниковой недостаточности и поражения спинного мозга и периферических нервов. У пациента снижается чувствительность конечностей, нарушаются процессы мочеиспускания, дефекации, снижается потенция. Позже развиваются эмоциональные нарушения, депрессия, недостаточность яичек, облысение.
У гетерозиготных женщин симптоматика проявляется к 40-60 годам. В тяжелых случаях наблюдаются церебральные симптомы, для умеренного течения болезни характерны симптомы адреномиелонейропатии с поражением нижних конечностей и расстройством функции тазовых органов. Надпочечниковая недостаточность наблюдается крайне редко.
причины
Причиной HDLs в большинстве семей мутации в колониестимулирующий фактор 1 рецептор (CSF1R), фактор роста для микроглии и моноцитов / макрофагов, предполагая , что микроглии дисфункция может быть первичным в ЛВП.
Мутации сконцентрированы в тирозин киназы области (Таэквон) белка. Мутации в основном были найдены в экзонах 12-22 в внутриклеточном ТКДЕ, в том числе 10 миссенса мутация , которые имеют одну нуклеотидную делецию и один кодон удаления , который состоит из триплета нуклеотидов , которые были удалены в результате чего в целом аминокислоты , чтобы не быть закодирована. Кроме того, три сплайсинга мутации сайта были идентифицированы , что вызвало в рамке удаление из в экзоне , выраженной нуклеотидной последовательности, что приводит к удалению более чем 40 аминокислот в ТКД.
Это определение имеет на основании генетических исследований 14 HDLs семей , подтверждающих мутации в этом гене. Белка рецептора CSF1 в основном функции в регуляции, выживания, пролиферации и дифференциации клеток микроглии. Механизм микроглии дисфункции из — за мутаций в CSF1R к потере миелина и аксонов сфероида формирования остается неизвестным. Необходимы дальнейшие исследования , чтобы лучше понять заболевания патогенез .
патогенез
Точная причина для разнообразной коллекции симптомов , найденной в различных ALD фенотипов не ясно. Белое вещество мозга, то клетки Лейдига этих яичек и коры надпочечников являются наиболее сильно пораженные системы. Избыток VLCFA может быть обнаружен почти во всех тканях организма, несмотря на локализацию симптомов. Отсутствие коэнзима А не допускает распад VLCFA, накапливая же в белом веществе надпочечников и семенников более конкретно в клетках Лейдига , не позволяя надлежащее функционирование этих органов. Успешное лечение процесса демиелинизации , который влияет на мозг либо с трансплантацией стволовых клеток или генной терапии не сразу нормализовать уровни VLCFA в тканях тела. Уровни VLCFA могут быть нормализованы путем обработки нефти Лоренца , но это не влияет на прогрессирование заболевания. Пока неясно , является ли накопление VLCFA связан с патогенезом заболевания определенным образом, или если он является биохимическим фенотипом, полезно для идентификации.