Способы лечения
В настоящее время медики и ученые всего мира не смогли разработать радикальные варианты избавления от описываемого заболевания. Поэтому для снижения неприятных ощущений у людей с ШМТ применяется симптоматические методики. Больному делают курсы внутримышечного введения витаминов группы В и Е. Для повышения состояния трофики используется АТФ и прочие вещества. Пациентам предписываются ингибиторные препараты холинэстеразы, фармакологические средства для повышения микроциркуляции, особый вид кислот и прочие лекарства. Помимо разных медикаментов больным рекомендуется проходить физтерапевтические процедуры. К ним относятся:
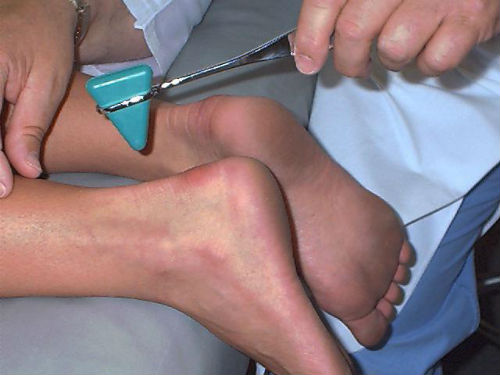
- Электрофорез — при которой организм человека подвергается влиянию электрических сигналов.
- Амплипульстерапия.
- Диадинамотерапия
- Грязелечение.
- Ультразвуковое лечение.
- Оксигенобаротерапия.
- Водолечение в сероводородных, сульфидных, хвойных, радоновых ваннах.
 Большое значение для выздоровления человека носит сохранение регулярных двигательных нагрузок, предотвращение формирования изменений и прочих аномалий.
Большое значение для выздоровления человека носит сохранение регулярных двигательных нагрузок, предотвращение формирования изменений и прочих аномалий.
Именно для такого используется ЛФК и массажные сеансы.
При острой необходимости врач-ортопед может предписывать разные ортопедические приемы.
Лечение болезни Шарко-Мари-Тута
Пока нет никакого лечения для ШМТ, но можно облегчить симптомы и отсрочить начало инвалидности.
НПВС (нестероидные противовоспалительные препараты), такие как ибупрофен, уменьшают суставные и мышечные боли, а также боли, вызванные поврежденными нервами.
Трициклические антидепрессанты (ТЦА) назначают, если НПВС не эффективны. ТЦА обычно используют для лечения депрессии, но они могут уменьшить болевые симптомы невропатии. Тем не менее, они имеют побочные эффекты.
Физическая терапия поможет укрепить и растянуть мышцы. Упражнения помогут сохранить мышечную силу.
Трудотерапия может помочь пациентам, которые имеют проблемы с движениями пальцев и им трудно осуществлять повседневную деятельность.
Ортопедические устройства могут предотвратить травмы. Обувь с высокими голенищами или специальные ботинки обеспечивают дополнительную поддержку голеностопного сустава, и специальная обувь или стельки для обуви могут улучшить походку.
Операция по удалению ахиллова сухожилия иногда может облегчить боль и сделать ходьбу легче. Хирургия может исправить плоскостопие, облегчить боль в суставах.
Клиническая картина
Основным симптомом заболевания являются амиотрофии, которые
начинаются симметрично с дистальных отделов нижних конечностей. В первую
очередь поражаются разгибатели и абдукторы стопы, в результате чего стопа
свисает, появляется характерная походка — степпаж (от англ. steppere — трудовая
лошадь). Сгибатели стопы и приводящие мышцы поражаются позже. Атрофия мышц
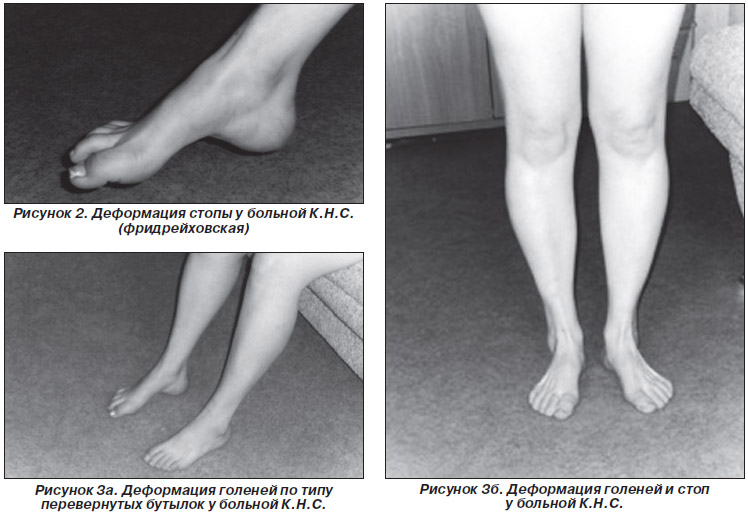
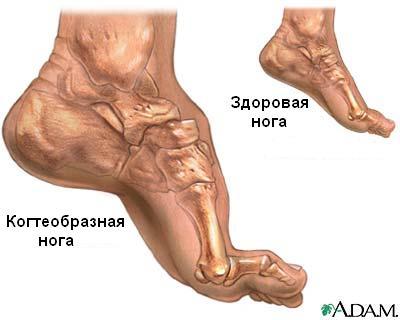
стопы приводит к когтевидной установке пальцев и деформации стопы, напоминающей
стопу Фридрейха. Амиотрофический процесс постепенно распространяется на более
проксимальные отделы. Однако в подавляющем большинстве случаев проксимальные
отделы конечностей остаются сохранными; процесс не распространяется также на
мышцы туловища, шеи и головы. При атрофии всех мышц голени образуется
болтающаяся стопа. На этой стадии болезни часто отмечается симптом «топтания»,
когда больные в положении стоя постоянно переминаются с ноги на ногу. Атрофия
мышц может распространяться на нижнюю часть бедер. Форма ноги в этих случаях
напоминает опрокинутую бутылку. Как правило, через несколько лет атрофии
распространяются и на верхние конечности. В первую очередь поражаются мелкие мышцы
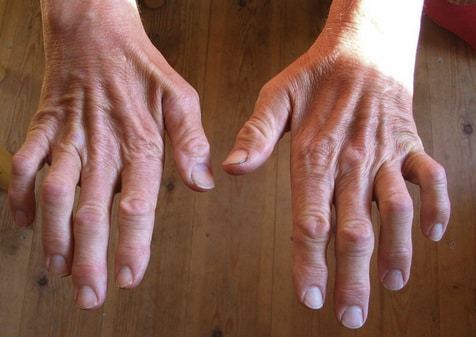
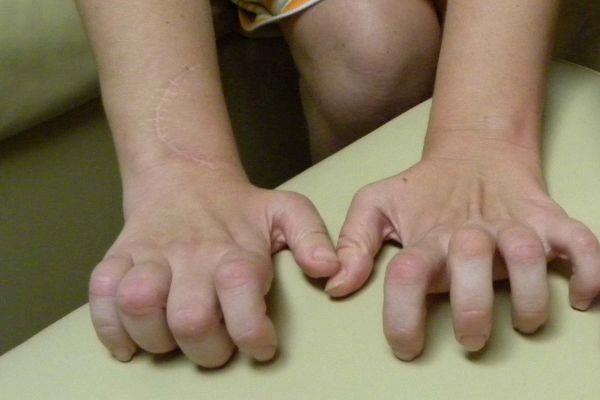
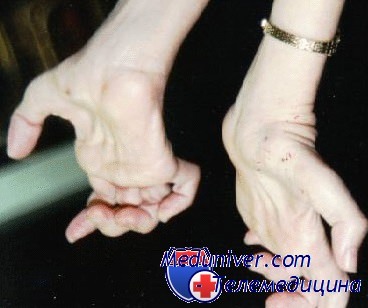
кисти, в результате чего кисть приобретает форму «обезьяньей лапы». Затем
в процесс вовлекаются мышцы предплечья. Мышцы плеча страдают в значительно
меньшей степени
Обращает на себя внимание то, что, несмотря на выраженные
атрофии мышц, больные могут в течение длительного времени сохранять
трудоспособность. При неавральной амиотрофии часто наблюдаются нередко
выраженные фасцикулярные подергивания в мышцах конечностей
При
электромиографическом исследовании выявляются признаки невритического,
переднерогового и супрасегментарного типов нарушения мышечного электрогенеза.
Мужчины болеют несколько чаще женщин. Заболевание обычно
начинается в детском возрасте — во второй половине первого или в первой
половине второго десятилетия жизни. Однако возраст начала болезни может широко
колебаться в разных семьях, что допускает возможность генетической
гетерогенности данного заболевания.
Течение болезни — медленно прогрессирующее. Между началом
амиотрофии в верхних и нижних конечностях может проходить до 10 лет и больше. Иногда
процесс обостряется в связи с различными экзогенными вредностями. В отдельных
случаях в течение длительного времени состояние больных может оставаться
стационарным.




Для невральной амиотрофии Шарко-Мари характерны также
нарушения чувствительности. В дистальных отделах конечностей определяется
гипестезия, причем поверхностные виды чувствительности, главным образом болевая
и температурная, страдают в значительно большей степени. Могут отмечаться боли
в конечностях, повышенная чувствительность к давлению нервных стволов.
В ряде случаев имеют место трофические нарушения — отек и
цианоз кожных покровов конечностей.
Клинические проявления заболевания в ряде семей могут
варьировать. Описаны семьи, где наряду с типичной невральной амиотрофией имели
место случаи гипертрофического полиневрита. В связи с этим некоторые авторы
объединяют эти заболевания в одну нозологическую форму.
Неоднократно подчеркивалась связь между невральной амиотрофией
и наследственной атаксией Фридрейха. Наблюдались семьи, у одних членов которых
имелась невральная амиотрофия, у других — атаксия Фридрейха. Описаны
промежуточные формы между этими заболеваниями; у отдельных больных типичная
клиническая картина атаксии Фридрейха через много лет сменялась картиной
невральной амиотрофии, которую отдельные авторы считают даже промежуточной
формой между атаксией Фридрейха и нейрофиброматозами.
Лечение
Лечение нейрогенных амиотрофии симптоматическое, комплексное
и пожизненное. Лечение и профилактика амиотрофии предполагают лечение основного
заболевания. Применяют витамины группы В, витамин Е, глутаминовую кислоту,
аминалон, прозерин, дибазол, биостимуляторы, антихолинэстеразные средства,
анаболические гормоны. При амиотрофиях, обусловленных заболеваниями, склонными
к регрессу, наряду с вышеуказанными средствами назначают электростимуляцию
периферических нервов, ванны, грязелечение. Периодически проводят курсы
анаболических стероидов, фармакотерапия. При нарушенной подвижности в суставах,
деформациях скелета больным необходима ортопедическая коррекция…
Online-консультации врачей
| Консультация радиолога (диагностика МРТ, КТ) |
| Консультация сурдолога (аудиолога) |
| Консультация психолога |
| Консультация невролога |
| Консультация хирурга |
| Консультация иммунолога |
| Консультация косметолога |
| Консультация диетолога-нутрициониста |
| Консультация уролога |
| Консультация эндокринолога |
| Консультация гинеколога |
| Консультация специалиста по лазерной косметологии |
| Консультация диагноста (лабораторная, радиологическая, клиническая диагностика) |
| Консультация психоневролога |
| Консультация доктора-УЗИ |
Новости медицины
Футбольные фанаты находятся в смертельной опасности,
31.01.2020
«Умная перчатка» возвращает силу хвата жертвам травм и инсультов,
28.01.2020
Назван легкий способ укрепить здоровье,
20.01.2020
Топ-5 салонов массажа в Киеве по версии Покупон,
15.01.2020
Новости здравоохранения
Глава ВОЗ объявил пандемию COVID-19,
12.03.2020
Коронавирус атаковал уже более 100 стран, заразились почти 120 000 человек,
11.03.2020
Коронавирус атаковал 79 стран, число жертв приближается к 3200 человек,
04.03.2020
Новый коронавирус атаковал 48 стран мира, число жертв растет,
27.02.2020
Развитие заболевания
Достоверной информации о том, что является причиной мутации и как происходит развитие заболевания, нет. Проведенные исследования дали понять, что от 70 до 80 % случаев, которые были обследованы генетически, наблюдалось дублирование некоторого участка 17-й пары хромосом. Установлено точно, что несколько форм заболевания имеют в своей природе мутации генов. Примером может стать нарушение кодирования гена MFN2, он отвечает за кодирование митохондриального белка. В результате образуется сгусток митохондрий (органелла клетки, отвечающая за образование энергии), который и препятствует нормальному проведению импульса.
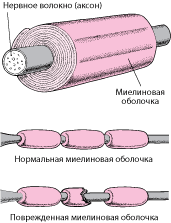
Также установлено, что при заболевании повреждается миелиновая оболочка периферических нервов, в более редких случаях можно встретить нарушения со стороны аксонов (это осевые цилиндры, которые проходят в центре нервного волокна). Процессы разрушения могут затронуть передние и задние спинномозговые корешки, клетки передних рогов, проводящие пути Голля (отвечают за проведение глубокой чувствительности по телу).
Вторичным явлением разрушения нервной оболочки становится атрофия мышечной ткани, она может затрагивать отдельные участки. Дальнейшее прогрессирование заболевания приводит к тому, что развивается замена мышечной ткани на соединительную.
Симптомы
Клиническая картина болезни имеет общие симптомы независимо от типа, но при этом может проявляться индивидуально. Даже в одной семье, когда заболевание провоцируется одним и тем же геном, у двух близких родственников оно далеко не всегда проявляется одинаково.
Общие симптомы болезни Шарко:
- атрофия мышц дистальных отделов конечностей (голень, предплечье);
- появление нечувствительных участков, онемение (без «мурашек» и покалываний);
- нарушение рефлексов;
- увеличение свода стопы (утолщение голеностопного сустава);
- сколиоз, кифоз, другие деформации скелета.
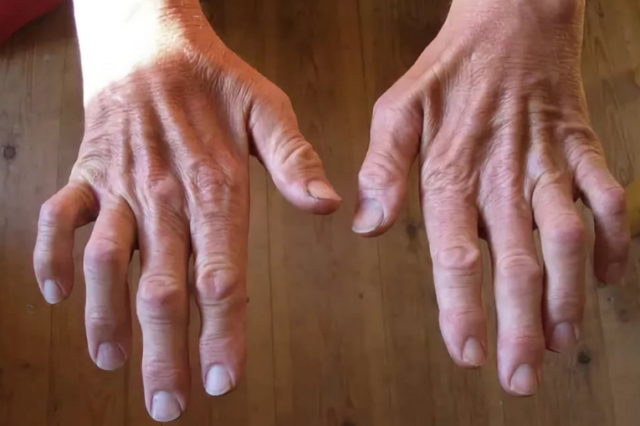
Дополнительные симптомы при 1-м типе болезни:
- сильные судороги в мышцах голени (чаще в передних отделах) после физической нагрузки;
- нарастающая слабость мышц;
- изменение походки, у детей – ходьба на цыпочках;
- нарушение тонуса мышц-сгибателей и разгибателей;
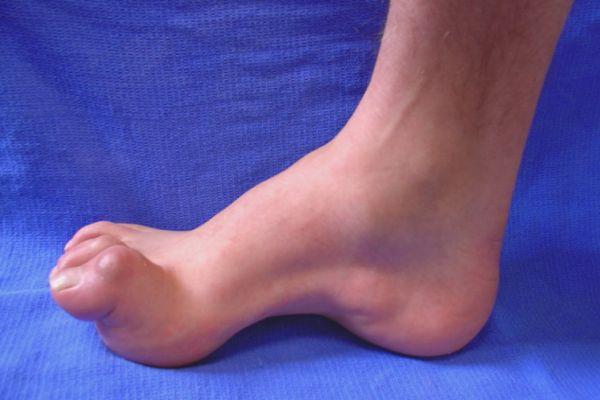
- молоткообразная форма пальцев ног (вместе с увеличением свода стопы);
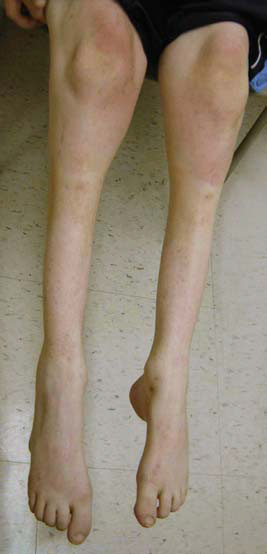
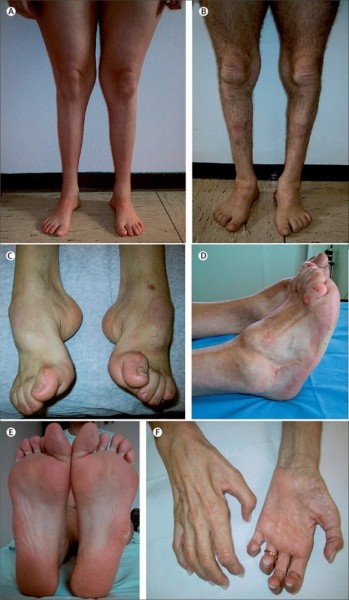
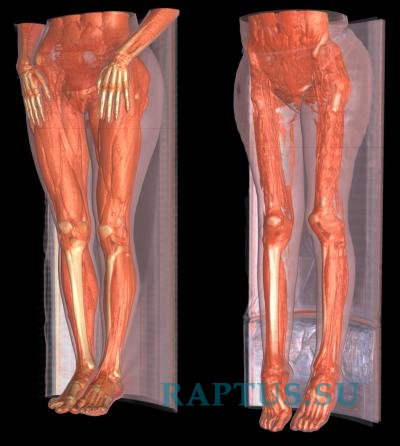
- атрофия мышц ног, начиная с дистальных (дальних) отделов – от стопы к колену; «аистообразная», «бутылкообразная» форма ног;
- по мере развития болезни – дрожание рук, слабость пальцев, нарушение мелкой моторики;
- нарушение сухожильных и периостальных рефлексов в области кистей и стоп;
- нарушение вибрационной, тактильной, а затем термальной и болевой чувствительности кистей и стоп;
- утолщение нервных стволов;
- искривления позвоночника;
- возможна атипичная форма болезни в виде синдрома Руси-Леви: топтание при попытке стоять неподвижно, неустойчивость при ходьбе, сильное дрожание рук в статичном положении, ночные судороги в икроножных мышцах, парезы.
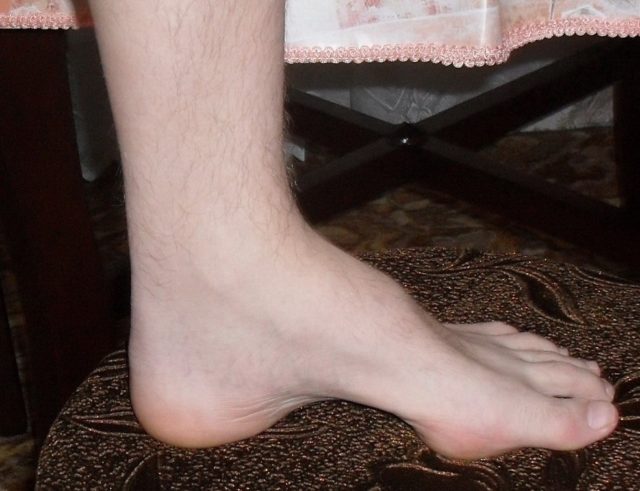
Особенности 2-го типа заболевания:
- изменения подъема стопы и формы пальцев встречаются реже, чем при 1-м типе;
- не наблюдаются утолщения нервных стволов;
- меньшая степень нарушения чувствительности;
- наблюдается синдром беспокойных ног перед сном;
- реже встречается ослабевание мышц в руках;
- возможно нарушение слуха (когда болезнь передается по женской линии);
- транзисторная энцефалопатия после физической активности на высоте: нарушения речи, шатание при ходьбе, затрудненное глотание, слабость проксимальных (ближних к туловищу) отделах конечностей.
Проявлением данной патологии обоих типов могут стать аутоиммунные реакции, когда вырабатываемые организмом специальные антитела разрушают миелиновые оболочки нервных волокон.
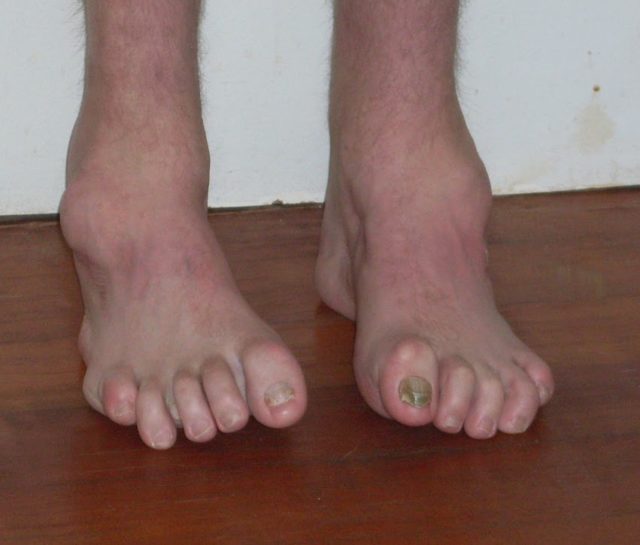
В чем замечены проявления невральной амиотрофии Шарко-Мари-Тута?
При описываемом заболевании у человека первым делом берет начало формирование схожих мышечных некрозов в нижних конечностей. Первые проявления, как правило, происходят такое в возрасте двадцати лет (намного реже, врачи могут диагностировать симптоматику в подростковый период от 16 лет, а также до 30 лет). Подобные проявления состоят в основном в высокой степени утомляемости ног во время долгого нахождения в вертикальном положении. Замечен синдром так называемого «топтания», то есть для того, чтобы пациент мог снять дискомфортные ощущения, он начинает ходить на месте. В некоторых редких ситуациях НА проявляется расстройстве чувствительности тех же отделов, чаще всего — образуются парестезии в виде ползания мурашек.
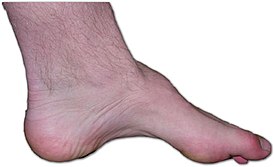




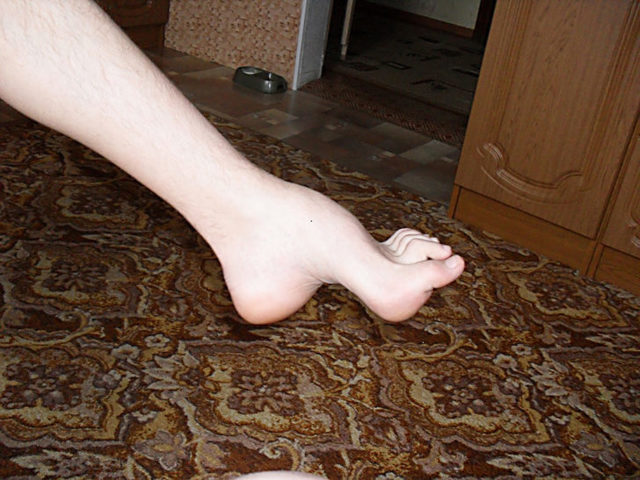



 Типичным ранних звоночком ШМ, считается полное отсутствие ахилловых и коленных рефлексов сухожилий. В итоге у больного проявляется свисание свода, неспособность ходить на пятках и анормальная походка, которая схожа с лошадиной. Впоследствии пагубный процесс увеличивает степень прогрессирования дальше и затрагивает мышцы и сгибатели. Максимальная уровень омертвения может привести к полной деформации ног с высоким сводом, схожу на тип стопы Фридрейха. Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Типичным ранних звоночком ШМ, считается полное отсутствие ахилловых и коленных рефлексов сухожилий. В итоге у больного проявляется свисание свода, неспособность ходить на пятках и анормальная походка, которая схожа с лошадиной. Впоследствии пагубный процесс увеличивает степень прогрессирования дальше и затрагивает мышцы и сгибатели. Максимальная уровень омертвения может привести к полной деформации ног с высоким сводом, схожу на тип стопы Фридрейха. Постепенно ШМТ переходит на проксимальные участки, к которым относятся голени и нижние зоны бедер. У пациента начинают серьезные деформации: свисающие стопы, ноги становятся нестандартной формы. Далее поражаются руки, кисть становится похожа на обезьянью лапу.
Стоит знать, что пагубные поражения никогда не поражают мышцы шейного периметра, туловища и плечевого района. Помимо вышеперечисленных симптомов, у больного могут появиться следующие проблемы:
- Слабое подергивание.
- Гипертрофия компенсаторной природы мышечных областей.
- Сенсорные нарушения.
- Возможность появления цианоза и отечности на кожном покрове.
Для ШМТ типично медленное развитие симптоматики. Период развития процесса уменьшения в размерах ног и рук может занять до десяти лет. Даже при таких серьёзных деформациях тела больной долгое время способен сохранять работоспособность и нормально выполнять различные бытовые обязанности. Ускорителями симптомов могут стать следующие факторы:
- Попадание в тело инфекционных агентов.
- Долгое пребывание на холоде.
- Травмирования головы.
- Повреждения спины и спинного мозга.
- Нехватка витаминов в организме.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При
ШMT 1
мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга. Это приводит к мышечной слабости и потере чувствительности или онемению.
При
ШМТ 2
мутирующий ген влияет непосредственно на аксоны. Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
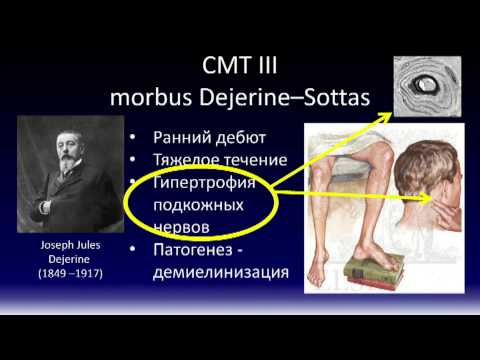
ШМТ 3
или
болезнь Дежерин-Соттас
, редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4
является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х
вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.
Как проявляется болезнь Шарко-Мари-Тута
Болезнь Шарко-Мари-Тута проявляется даже в пределах одной семьи не всегда одинаково. И дело не в многообразии её признаков. А в том, что гены, кодирующие данную патологию, способны с разной степенью выраженности формировать симптомы. Проще говоря, имея идентичную «поломку» в хромосомах, признаки недуга у отца и сына будут иметь индивидуальную окраску.
Общие симптомы
Клиническая картина заболевания практически не зависит от его типа и включает:
- атрофию мышц дистальных, то есть наиболее отдалённых от туловища, отделов конечностей;
- снижение сухожильных и периостальных рефлексов;
- изменение чувствительности, характеризующееся её выпадением, но никогда не сопровождающееся появлением ощущения покалывания или «ползания мурашек»;
- деформацию опорно-двигательного аппарата – сколиоз, увеличение свода стопы и др.
Но всё же существует ряд признаков несколько отличающих течение заболевания при различных его вариантах.
Первый тип
Болезнь Шарко-Мари-Тута первого типа нередко протекает в исключительно стёртой форме, при которой пациенты не ощущают изменений в организме и не обращаются за медицинской помощью вообще. Если же патология себя проявила, то происходит это в период первого, максимум второго, десятилетия жизни.
При этом наблюдаются:
- болезненные судороги в мышечном массиве голени, причём редко в икроножной мышце, чаще в передней группе мышц. Подобные спазмы нарастают после периода длительной физической нагрузки (ходьбы, занятий спортом, езде на велосипеде);
- изменения в походке, связанные с постепенным усилением слабости в мышцах. При этом у детей это может дебютировать в хождении на цыпочках;
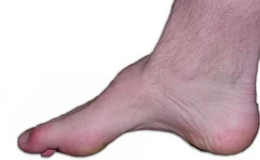
- деформация стоп с формированием высокого свода последних и наличием молоточкообразных пальцев, которая развивается как результат дисбаланса в тонусе сгибателей и разгибателей;
- атрофия мышц, начинающаяся со стоп и поднимающаяся на голень. Затем процесс затрагивает кисть – появляется тремор в руках и выраженная слабость в пальцах, особенно при попытке выполнять мелкие движения. К примеру, застёгивать пуговицы, перебирать крупу;
- угнетение или полное отсутствие сухожильных и периостальных рефлексов, а именно ахиллового, карпорадиального, при сохранных с более проксимальных отделов рук и ног. То есть коленный и рефлексы с двуглавой и трёхглавой мышцы остаются интактными;
- нарушения чувствительности в кистях и стопах, выражающиеся в её постепенном выпадении. Причём стартует патология с вибрационной и тактильной сферы, распространяясь на суставно-мышечные и болевые ощущения;
- сколиоз и кифосколиоз;
- утолщение нервных стволов, чаще всего поверхностного малоберцового и большого ушного.
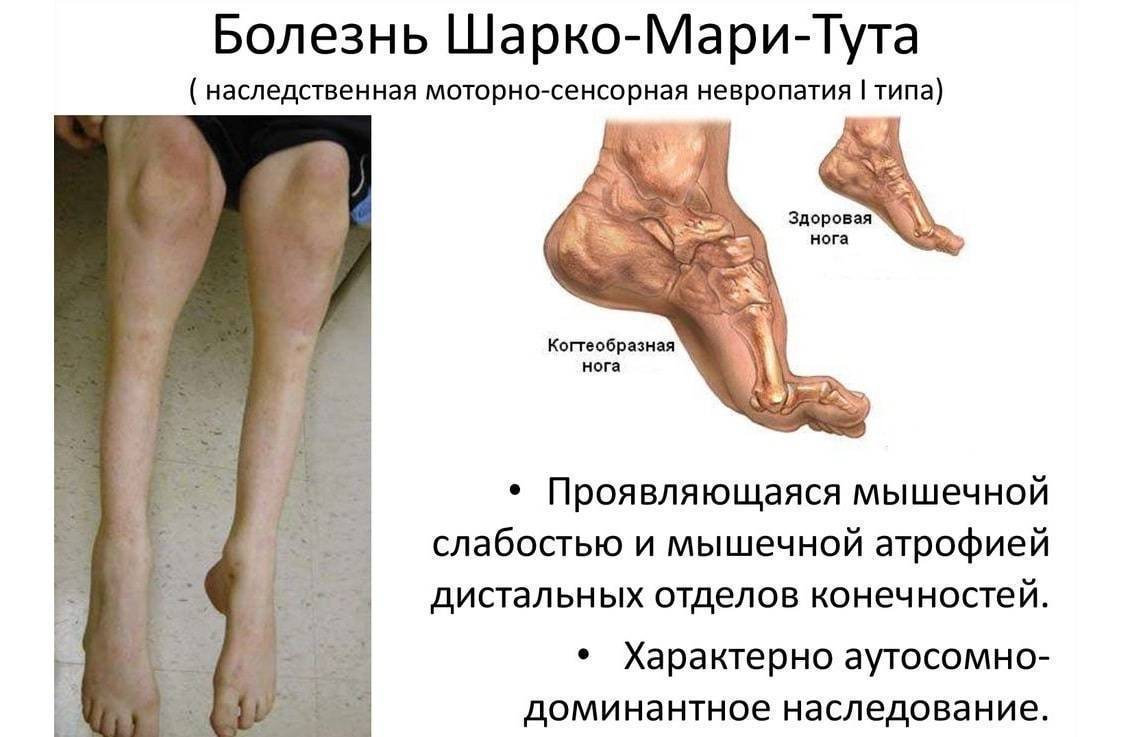
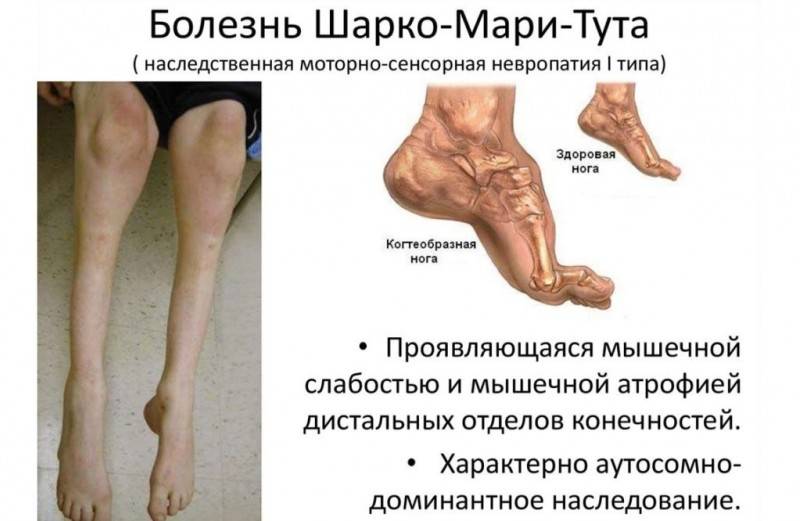

Для болезни Шарко характерна атрофия мышц именно в дистальных отделах конечностей. При этом если не выражена подкожно-жировая клетчатка, объём голени и бедра разительно отличается, и ноги приобретают вид таковых у аиста или походят на перевёрнутую бутылку для шампанского.
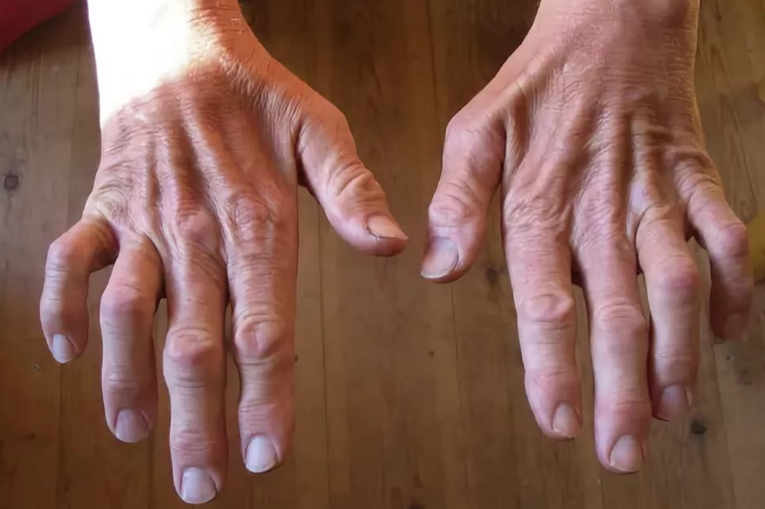
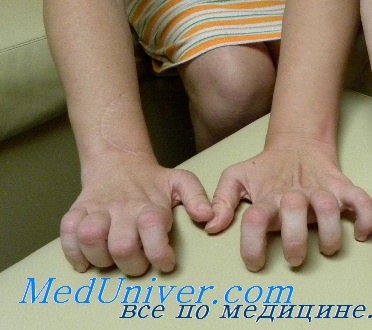





Невральная амиотрофия Шарко Мари первого типа имеет атипичные формы. Одна из них — синдром Руси-Леви, при котором наблюдается выраженный тремор при попытке удержать руки в одном положении и неустойчивость при ходьбе. Сюда же относится заболевание, проявляющееся, помимо стандартных симптомов, парезами, гипертрофией мышц голени, резким выпадением чувствительности и ночными судорогами в икроножных мышцах.
Второй тип
Для болезни Шарко-Мари-Тута второго типа, кроме более позднего начала, характерны:
- менее выраженные изменения чувствительности;
- более редкая встречаемость деформаций стопы и пальцев;
- наличие синдрома беспокойных ног (возникают неприятные ощущения в ногах во время отхода ко сну, заставляющие пациента двигаться, что облегчает состояние);
- нередко сохранная сила в кисти;
- отсутствие утолщения нервных стволов.
При синдроме Шарко-Мари-Тута, передающемся через Х-хромосому, могут встречаться нейросенсорная тугоухость (снижение слуха) и транзиторная энцефалопатия, возникающая после физической нагрузки на высоте. Для последней характерно появление симптомов через 2-3 дня после занятий. Признаками патологии становятся шаткость, нарушение речи, глотания, слабость в проксимальных отделах рук и ног. Обычно клиническая картина недуга исчезает самостоятельно в течение пары недель.
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.



- (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
- (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Наследственное заболевание. Основной тип передачи – аутосомно-доминантный (с пенетрантностью патологического гена около 83%), реже – аутосомно-рецессивный.
Морфологическую основу болезни составляют дегенеративные изменения главным образом в периферических нервах и нервных корешках, касающиеся как осевых цилиндров, так и миелиновой оболочки. Иногда наблюдаются гипертрофические явления в интерстициальной ткани. Изменения в мышцах носят преимущественно неврогенный характер, отмечается атрофия отдельных групп мышечных волокон; в неатрофированных мышечных волокнах структурные изменения отсутствуют. По мере прогрессирования заболевания появляются гиперплазия интерстициальной соединительной ткани, изменения в мышечных волокнах – их гиалинизация, центральное смещение ядер сарколеммы, гипертрофия некоторых волокон. В более поздних стадиях болезни отмечаются гиалиновая дегенерация, распад мышечных волокон. Наряду с этим в ряде случаев отмечены изменения в спинном мозге. Они складываются из атрофии клеток передних рогов, главным образом в поясничной и шейной части спинного мозга, и различной степени поражения проводниковых систем, характерного для наследственной атаксии Фридрейха.
Осложнения
Если запустить невральную амиотрофию, то могут быть необратимые последствия. Проявляются они в виде нарушения дыхательной системы. Если патология поражает нервные окончания, которые контролируют диафрагму. Больному скорее всего нужно будет употреблять бронхолитические средства или искусственную вентиляцию легких. Лишний вес или сильное ожирение может привести к тому, что пациенту становится трудно дышать.
Депрессивное состояние может быть из-за стрессовых ситуаций. Чаще всего возникает это из-за прогрессирования патологии. Могут применяться, как специальные лекарственные препараты, так и психологическая помощь. Если заболевание будет в запущенной форме и без лечения, то это приведет к инвалидности.

Пациент может перестать полностью передвигаться и на этой фоне возникнуть глухота. Для того чтобы не было такие серьезных последствий, необходимо своевременно обращаться к врачу. Он сможет назначить комплексную диагностику, чтобы поставить точный диагноз. Только после этого будут применяться методы лечения. Заболевание предоставляет хроническую моторную и сенсорную полинейропатию.
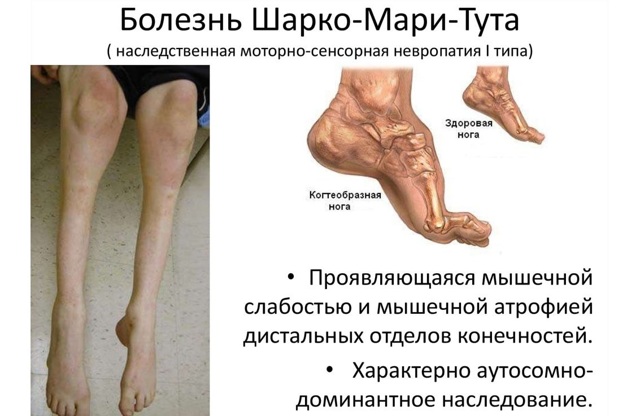
1.2.2. Невральная амиотрофия шарко-мари-тута.
Частота 1:500000 населения. Наследуется по аутосомно-доминантному, аутосомно-рецессивному сцепленному с Х-хромосомой типу. Обнаруживается сегментарная демиелинизация в нервах, в мышцах — денервация с явлениями «пучковой» атрофии мышечных волокон.
КЛИНИКА. Первые признаки заболевания чаще проявляются в 15 — 30 лет, реже в дошкольном возрасте. Характерными симптомами являются мышечная слабость, патологическая утомляемость в дистальных отделах ног. Больные быстро устают при длительном стоянии на одном месте и нередко для уменьшения утомления в мышцах прибегают к ходьбе на месте («симптом топтания«). Реже заболевание начинается с чувствительных расстройств — болей, парестезий, ощущения ползания мурашек. Атрофии первоначально развиваются в мышцах голеней и стоп. Мышечные атрофии, как правило, симметричные. Поражается перонеальная группа мышц и передняя большеберцовая мышца. Вследствие атрофий ноги резко сужаются в дистальных отделах и приобретают форму «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся «выеденными» с высоким сводом. Парез стоп изменяет походку больных. Они ходят, высоко поднимая ноги; ходьба на пятках невозможна. Атрофии в дистальных отделах рук — мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются позже. Ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлекс с трех-двуглавой мышц плеча длительное время остаются сохранными. Чувствительные расстройства объективно определяются нарушениями поверхностной чувствительности по периферическому типу (тип «перчаток» и «носков»). Часто имеются вегетативно-трофические нарушения — гипергидроз стоп и кистей, гиперемия кистей и стоп. Интеллект обычно сохранен.
Течение медленно прогрессирующее. Прогноз благоприятен.
Диагностика. Тип наследования, атрофии дистальных отделов конечностей, расстройства чувствительности по полиневритическому типу, медленное прогрессирующее течение, результаты электромиографии (снижение скорости проведения по периферическим нервам), биопсия нервов.
ЛЕЧЕНИЕ ПМД. Применяют витамины группы В, С Е, а также АТФ, церебролизин, ноотропил, энцефабол, фосфаден, карнитина хлорид, метионин, лецитин, глутаминовая кислота, ретаболил. Положительный эффект дают антихолинэстеразные препараты (прозерин, местинон, галантамин). Показаны средства, улучшающие микроциркуляцию: никотиновая кислота, трентал, пармидин. Наряду с медикаментозной терапией применяют ЛФК, массаж, электрофорез лекарственных средств (прозерин, кальция хлорид), диадинамические токи, синусоидальные модулированные токи, электростимуляция, ультразвук, озокерит, грязевые аппликации, радоновые, хвойные, сульфидные и сероводородные ванны, оксигенобаротерапия. Показано ортопедическое лечение при контрактурах конечностей, умеренной деформации позвоночника и асимметричном укорочении конечностей.





13.2. Семейная атаксия фридрейха.
Наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых столбов спинного мозга. Тип наследования аутосомно-рецессивный с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
КЛИНИКА. Начало заболевания относится к 6 — 15-летнему возрасту. Первым симптомом болезни является неустойчивая походка. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на руки и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Нарушается почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередко патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен. Заболевание медленно прогрессирует. Средняя продолжительность жизни 10 — 15 лет с момента его развития.
Диагностика. Заболевание распознается на основании характерных симптомов — деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы, флексия концевых фаланг), позвоночника, поражения миокарда, эндокринных расстройств.
ЛЕЧЕНИЕ. Применяются симптоматические средства: общеукрепляющие препараты, ЛФК, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Спинальные амиотрофии
Спинальная амиотрофия представляет собой прогрессирующее заболевание в результате которого поражаются нервные клетки спинного мозга. Это не одно заболевание, сюда входит целая группа болезней: болезнь Арана-Дюшена, болезнь Вердинга-Гоффмана и ряд других более редких заболеваний.
Несмотря на многочисленность заболеваний, входящих в данную группу, все они проявляются сходными симптомами. Это выражается в том, что со временем развиваются вялые параличи, ослабевают сухожилия. Как правило, поражения ассиметричны. Особенности каждого заболевания в том, что страдают вначале различные группы мышц.
Например, при заболевании Вердинга-Гоффмана у больного наблюдается слабость, в основном страдают мышцы туловища. Исследователи отмечают высокий процент кровного родства среди родителей заболевших. Данное заболевание подразделяется на виды в зависимости от времени появления и прогрессирования заболевания: врожденная, ранняя детская и поздняя.
Развитие врожденной амиотрофии происходит в первые месяцы жизни малыша. Данное заболевание, как правило, сочетается с другими пороками. При запоздалом лечении высока вероятность летального исхода. Причиной последнего является сердечнососудистая и дыхательная недостаточность, которая развивается из-за слабости дыхательных мышц.
Ранняя детская амиотрофия развивается в возрасте от полугода до одного года. Изначально поражаются мышцы туловища и ног, в дальнейшем происходит нарушение работы всех групп мышц. Обнаружить это заболевание достаточно просто.
Малыш не встает на ножки, не может сидеть и с трудом захватывает игрушки. Характерной чертой также является небольшое подергивание мышц, особенно языка. Если вовремя не приступить к лечению, то развивается полная гипотония мышц и паралич. Ребенок с таким заболеванием не доживает до 15 лет.
Поздняя амиотрофия проявляется в возрасте от двух с половиной до трех с половиной лет. В это время ребенок уже устойчиво стоит на ногах и свободно передвигается в пространстве.
Симптомами этого заболевания является неуверенность при ходьбе и частые падения. Данное заболевание постепенно прогрессирует, поражая все большую группу мышц. В итоге к десяти годам ребенок перестает самостоятельно передвигаться и не может себя обслуживать. Прожить с подобным заболеванием человек может лишь максимум до 30 лет.
Доброкачественная спинальная амиотрофия Кугельберга-Веландер. Отдельное заболевание, которое входит в группу болезней спинальной амиотрофии. Отдельная группа исследователей считает, что данное заболевание является разновидностью болезни Вердинга-Гоффмана.
Данное заболевание медленно прогрессирует, развивается, как правило, в мышцах туловища и постепенно распространяется на конечности. Сопровождается общей слабостью. Наблюдается у детей в возрасте от трех до семнадцати лет. Характерной чертой данной болезни является также избыточная масса тела. Люди с подобным заболеваниям доживают до глубокой старости и сохраняют способность самостоятельно передвигаться.
Болезнь Арана-Дюшера наблюдается у лиц в старческом возрасте. Характеризуется ослаблением мышц конечностей. Само течение болезни медленное. Наблюдается подергивание мышц, а в отдельных случаях и параличи. Смерть при данном заболевании наступает от бронхопневмонии.
Признаки амиотрофии невральной Шарко-Мари
Характерным и ранним признаком болезни является отсутствие или значительное снижение сухожильных рефлексов. В первую очередь исчезают ахилловы, а затем коленные рефлексы. Однако в отдельных случаях могут иметь место повышение сухожильных рефлексов, патологический симптом Бабинского. Эти признаки, связанные с поражением боковых столбов спинного мозга, наблюдаются только в ранних стадиях или при рудиментарных формах болезни. В проксимальных отделах конечностей может иметь место компенсаторная гипертрофия мышц.
Для невральной амиотрофии характерны также нарушения чувствительности. В дистальных отделах конечностей определяется гипестезия, причем поверхностные виды чувствительности, главным образом болевая и температурная, страдают в значительно большей степени. Могут отмечаться боли в конечностях, повышенная чувствительность к давлению нервных стволов.
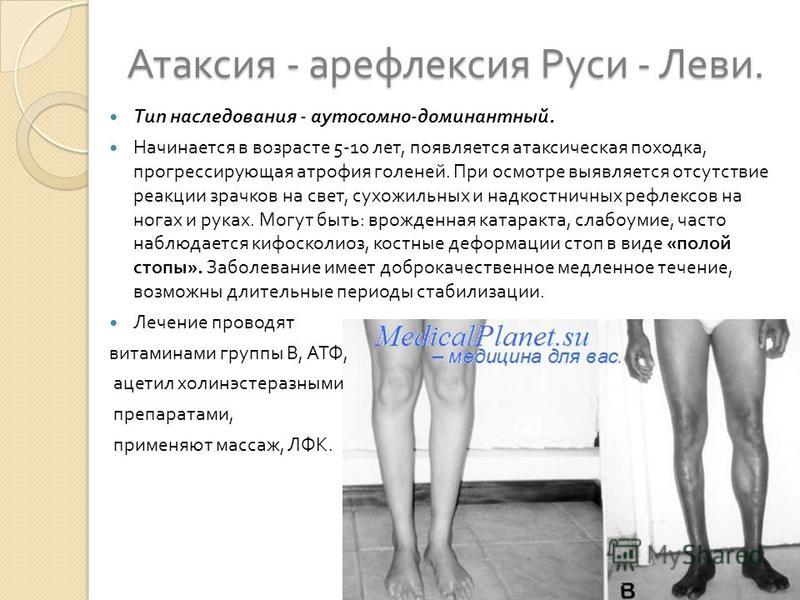
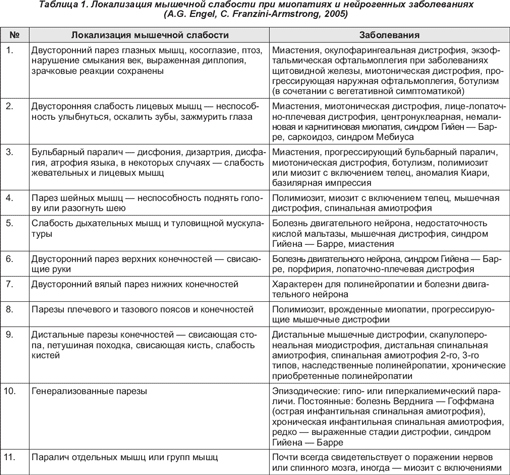



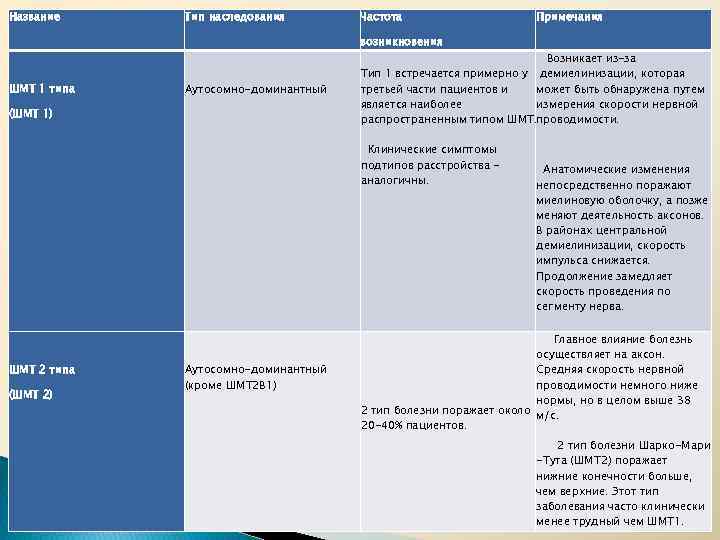
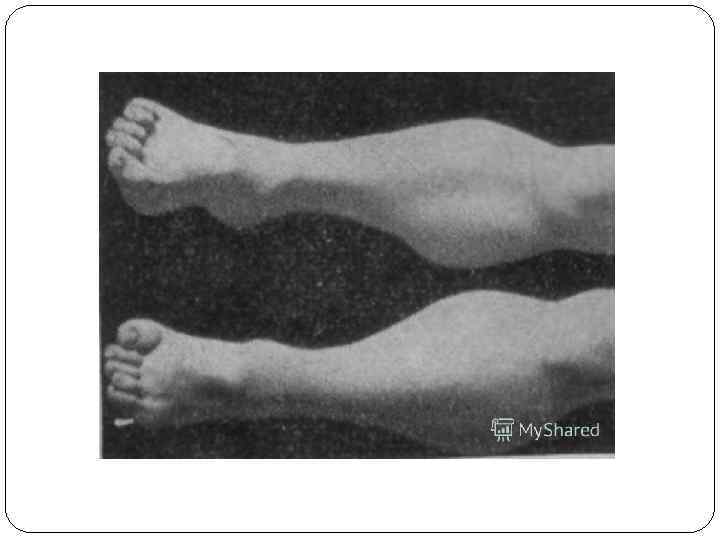
В ряде случаев имеют место трофические нарушения – отек и цианоз кожных покровов конечностей.
Клинические проявления заболевания в ряде семей могут варьировать. Описаны семьи, где наряду с типичной невральной амиотрофией имели место случаи гипертрофического полиневрита. В связи с этим некоторые авторы объединяют эти заболевания в одну нозологическую форму.
Неоднократно подчеркивалась связь между невральной амиотрофией и наследственной атаксией Фридрейха. Наблюдались семьи, у одних членов, которых имелась невральная амиотрофия, у других – атаксия Фридрейха. Описаны промежуточные формы между этими заболеваниями; у отдельных больных типичная клиническая картина атаксии Фридрейха через много лет сменялась картиной невральной амиотрофии, которую отдельные авторы считают даже промежуточной формой между атаксией Фридрейха и нейрофиброматозами.
Иногда наблюдается сочетание невральной амиотрофии с миотонической дистрофией.
Мужчины болеют несколько чаще женщин. Заболевание обычно начинается в детском возрасте — во второй половине первого или в первой половине второго десятилетия жизни. Однако возраст начала болезни может широко колебаться в разных семьях, что допускает возможность генетической гетерогенности данного заболевания.
Течение болезни — медленно прогрессирующее. Между началом амиотрофии в верхних и нижних конечностях может проходить до 10 лет и больше. Иногда процесс обостряется в связи с различными экзогенными вредностями. В отдельных случаях в течение длительного времени состояние больных может оставаться стационарным.
Невральную амиотрофию иногда трудно дифференцировать от различных хронических полиневритов, при которых также наблюдаются дистальные атрофии мышц. В ее пользу говорят наследственный характер и прогрессирующее течение болезни. От дистальной миопатии Гоффманна невральная амиотрофия отличается фасцикулярными подергиваниями в мышцах, нарушениями чувствительности, отсутствием поражения мышц туловища и проксимальных отделов конечностей, а также электромиографической картиной.
Гипертрофический интерстициальный неврит Дежерина — Сотта отличается от невральной амиотрофии значительным утолщением (часто узелковым) нервных стволов, атаксией, сколиозом, более грубыми изменениями болевой чувствительности, частым присутствием зрачковых нарушений, нистагмом.